

Guía de Práctica Clínica para las Distrofias Hereditarias de Retina
Completa HTML

5.1. Detección precoz
- En pacientes en edad pediátrica que acuden a la atención primaria (o a urgencias), ¿qué conjunto de signos y síntomas debe despertar sospecha de DHR?
- En pacientes adultos que acuden a la atención primaria (o a urgencias), ¿qué conjunto de signos y síntomas debe despertar sospecha de DHR?
No se ha encontrado evidencia científica que responda a estas preguntas de forma directa. La respuesta se ha basado en el documento elaborado por la Sociedad Española de Retina y Vítreo para el diagnóstico y manejo de las enfermedades hereditarias de retina.18
Edad Pediátrica
Aunque las enfermedades de la retina en edad pediátrica son poco frecuentes, gran parte de las DHR tienen su inicio en esta edad. Puesto que los niños hasta los 5-6 años raramente se quejan de pérdida de visión, toman especial relevancia los signos indirectos en los niños más pequeños (como notar que el niño se acerca mucho a los objetos, desviaciones oculares, reflejo blanco en la pupila, etc.), y la AV, que los pediatras deben tomar a partir de los tres años de edad, pues ambos pueden orientar hacia la sospecha de que algo anómalo está pasando, lo que incluye el inicio de estas patologías. Además, debe ser remitido al oftalmólogo cualquier niño que no mejora su visión con gafas. Muy específicamente hay que estar atento a si tiene sintomatología específica de RP, como puede ser que se tropiece con mucha frecuencia (debido a los problemas de campo) o que tenga dificultades a la hora de desplazarse por la oscuridad.
muy baja
Se enviará al niño a oftalmología siempre que haya sospecha de que la visión no es normal. En un recién nacido o un lactante se consideran signos de alteración en el desarrollo visual: movimientos erráticos de los ojos; no fija ni sigue objetos de colores o luces brillantes; no responde a caras familiares como la de su madre; nistagmus (el nistagmus secundario a déficit de información visual aparece a los 2-3 meses); mira hacia luces muy intensas sin prestar atención a ninguna otra cosa; se frota los ojos (reflejo óculo-digital signo de Franceschetti que sugiere deficiencia visual muy severa) lo que es muy típico de la ACL.
muy baja
La valoración de la AV de un niño más mayor se realiza en los controles pediátricos rutinarios con diferentes test según la edad:
- 2-3,5 años: Test de dibujos (Pigassou): Dibujos sencillos (árbol, casa, coche, niño, pájaro, sol)
- 3,5-5 años: Optotipos E de Snellen (el niño puede colocar una E de cartulina negra hacia dónde está abierta sin pedirle que diga derecha-izquierda-arriba-abajo, algo de lo que sería incapaz por sus problemas de lateralidad hasta los 5 años); o Optotipos C de Landolt (el niño tiene que identificar la dirección hacia la que se orienta la abertura de la C). Ambos son más fiables que el Pigassou.
- 5 años: Optotipos con números o letras, o bien E de Snellen o C de Landolt, igual que un adulto
Se considera que la visión del niño normal es:
- 1 mes: 5% de la visión del adulto (entre 20/200 y 20/400)
- 2-4 meses: 20% de la visión del adulto (entre 20/200 y 20/100)
- 1 año: 30-40% de la visión del adulto (entre 20/60 y 20/40)
- 3-4 años (incluso más tarde pues hay retrasos de maduración): 80-100% de la visión del adulto. Se consideraría sospechoso que el niño no viese más de 20/40
- Todo niño > 6-7 años debe ver igual que un adulto (AV=20/20) pero se consideraría patológico que no viese más de 20/32 y sobre todo si no mejora con gafas
Cualquier niño que se aleje de esta evolución normal debe ser remitido al oftalmólogo especialista en pediatría que valorará si enviarlo al experto en retina.
Se estima que el 16% de las DHR son sindrómicas, siendo las más frecuentes el USH (con RP + sordera, supone un 20% de las sindrómicas y responsable de un 6-10% de todas las sorderas neurosensoriales congénitas) y el Bardet-Biedl (9% de las sindrómicas con DHR, obesidad, polidactilia e hipogonadismo), ambas de herencia AR.18 Por tanto, en general, pero de modo especial en los pacientes pediátricos con enfermedad retiniana diagnosticada, se debe explorar la audición, el desarrollo psicomotor, la existencia de polidactilia (a menudo se ha operado antes del inicio de la consulta oftalmológica) y posibles problemas neurológicos o renales. Esta breve anamnesis dirigida puede ayudar a la orientación diagnóstica del caso.
muy baja
Edad adulta
Las DHR incluyen aquellas enfermedades que afectan fundamentalmente a la retina central (distrofias maculares) y aquellas que afectan a la visión periférica (las RP). Habrá que remitir a oftalmología a aquellos pacientes con historia familiar de enfermedad ocular de este grupo, sea cual sea la sintomatología ocular y sea cual sea el patrón de herencia ya que este es variable para este grupo de enfermedades. En general en las DHR es muy frecuente que los pacientes se quejen de fotofobia (la luz les molesta mucho) y de deslumbramiento importante (la visión baja cuando hay exceso de luz). También pueden referir problemas cuando pasan de luz a oscuridad (adaptación a la oscuridad) y viceversa, así como deslumbramiento indirecto (debido a reflejos transversales). Es sospechoso que el paciente se queje de mala visión cuando hay poca luz (ceguera nocturna) junto con ese empeoramiento de la visión si hay un exceso de luz (deslumbramiento). Este conflicto con la luz es típico de los pacientes con enfermedades de la retina en general, pero muy particularmente de las DHR.
muy baja
Los síntomas de las distrofias maculares son similares a los de cualquier maculopatía, con la peculiaridad de que la mayor parte de las maculopatías prevalentes se dan en personas de edad, por lo que la edad de presentación más temprana es importante para sospechar etiología hereditaria. Las DHR se diagnostican a menudo durante la niñez, la juventud o en edades medias de la vida y la progresión es gradual. El síntoma fundamental de las distrofias maculares es la disminución de la AV sin causa aparente (una vez descartadas las causas más frecuentes tales como errores refractivos o cataratas). Debido a la afectación central, los pacientes pueden referir síntomas del síndrome macular:
- Disminución de la AV, normalmente sin afectación del CV periférico
- Escotoma central (absoluto/relativo): manchas negras o grisáceas que no cambian de posición y en las que desaparecen los objetos del CV
- Discromatopsia: alteración de la visión de colores
- Metamorfopsia (en objetos cotidianos o con Rejilla Amsler)
- Deslumbramiento
muy baja
También hay que remitir a aquellos pacientes con sospecha de RP, es decir aquellos que refieran ceguera nocturna (se manejan muy mal en condiciones escotópicas) y/o reducción concéntrica del CV (visión en túnel); se puede comprobar en consulta con un CV por confrontación. A menudo los pacientes lo que refieren es que tropiezan demasiado al no ver los obstáculos que los rodean lo que dificulta su deambulación. En presencia de uno o ambos signos se puede empezar a sospechar una DHR18. Sin embargo, más adelante estos pacientes sufren disminución de visión (dificultades de lectura, dificultades para ver escalones), acompañada de alteración de la visión de colores. A veces, los pacientes van acostumbrándose a la afectación periférica que progresa muy lentamente, y sólo consultan cuando comienzan con la afectación central.
muy baja
Resumen de la evidencia
| Calidad muy baja | Signos indirectos tales como acercarse mucho a los objetos, desviaciones oculares, reflejo blanco en la pupila (leucocoria), frecuentes tropiezos o mal manejo en la oscuridad, y la pérdida de AV deben hacer sospechar al pediatra.18 |
| Calidad muy baja | Cualquier niño que no mejora su visión con su mejor corrección debe de ser remitido al oftalmólogo experto en retina.18 |
| Calidad muy baja | El pediatra debe remitir al oftalmólogo experto en oftalmología pediátrica a los niños en los que sospeche anomalías del sistema visual.18 |
| Calidad muy baja | Los síntomas y signos claves para el médico de familia en la sospecha de una DHR son la ceguera nocturna y reducción concéntrica del CV que a veces manifiestan como dificultades en la deambulación.18 |
| Calidad muy baja | Signos como la obesidad con polidactilia, el hipogonadismo, la discapacidad intelectual, la insuficiencia renal o cardíaca así como la sordera de origen neurosensorial bilateral pueden hacer sospechar al pediatra de atención primaria de la existencia de una DHR sindrómica.18 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- Calidad global de la evidencia: No se identificó evidencia científica disponible sobre qué conjunto de signos y síntomas debe despertar sospecha de DHR.
- Balance entre beneficios y riesgos: El grupo de elaboración de la guía opina que los beneficios de tener en cuenta los signos y síntomas que se apuntan superan el posible perjuicio para el paciente.
- Valores y preferencia de los pacientes: No se identificaron estudios publicados sobre valores y preferencias de las personas con DHR en cuanto a esta pregunta. En el estudio cualitativo realizado en el contexto español, no se pudo obtener información al respecto.
- Costes y uso de recursos: no se consideraron determinantes para formular las recomendaciones.
Las recomendaciones formuladas para esta pregunta son de buena práctica clínica y se tratan de directrices dirigidas a realizar una adecuada detección y un adecuado manejo de los primeros signos y síntomas asociados a las DHR, tanto en pacientes pediátricos como en adultos.
Recomendaciones
| √ | Se sugiere que el pediatra de atención primaria envíe al oftalmólogo a aquellos niños que refieran pérdida de visión. |
| √ | Se sugiere que el pediatra de atención primaria envíe al oftalmólogo a todo niño al que se detecte una visión no adecuada para su edad o sospecha de la misma en el caso de los más pequeños, vaya o no asociada a sintomatología sistémica. |
| √ | Se sugiere que los pediatras de atención primaria deriven al servicio de oftalmología (en caso de disponibilidad, preferentemente, al servicio de oftalmología pediátrica) a aquellos niños que presenten sordera bilateral, con o sin sospecha de pérdida de visión. |
| √ | Se sugiere que los pediatras de atención primaria deriven al servicio de oftalmología pediátrica a aquellos niños que asocien obesidad, polidactilia e hipogonadismo, y se sospeche de pérdida de visión. |
| √ | Se sugiere que los médicos de atención primaria deriven al servicio de oftalmología a aquellos pacientes con ceguera nocturna y reducción concéntrica del campo visual. |
| √ | Se sugiere que los médicos de atención primaria deriven al servicio de oftalmología a aquellos pacientes con disminución de la visión central. |
5.2. Confirmación diagnóstica
5.2.1. Pruebas diagnósticas
- En pacientes con sospecha de DHR, ¿qué pruebas se deben realizar para confirmar o descartar el diagnóstico? ¿Por qué orden? ¿A quién?
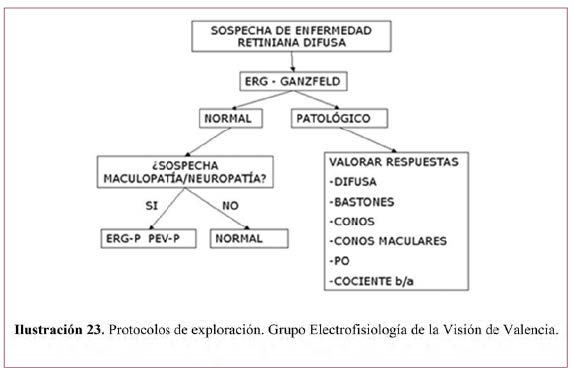
En pacientes con sospecha de DHR, dentro de las pruebas que se pueden realizar para confirmar el diagnóstico encontramos aquellas subjetivas, que normalmente requieren mayor colaboración de los pacientes y, por otro lado, las pruebas objetivas, algunas de ellas de fácil realización en pacientes no colaboradores como por ejemplo los niños.
La respuesta a esta pregunta clínica se ha basado en el documento de la Sociedad Española de Retina y Vítreo18 para las enfermedades hereditarias de retina y en varias publicaciones que recogen opiniones de expertos, una sobre el diagnóstico diferencial de las distrofia maculares hereditarias;70 otra sobre el diagnóstico genético y perfil molecular de pacientes con distrofia de retina en general en población española71 u otras poblaciones, otra sobre XLRS72 y otra más sobre imagen y electrofisiología clínica.73 Por otro lado, se identificaron 17 publicaciones en las que se recogían diversas cohortes, revisiones de casos y series de casos que incluyeron un total de 950 pacientes con diferentes DHR entre las que se encuentran la RP, coroideremia, el USH, las distrofias de conos y bastones, la distrofia foveomacular viteliforme, el síndrome de Stargardt o fundus flavimaculatus y la retinosquisis juvenil ligada al cromosoma X. Las pruebas diagnósticas incluidas son: AV y sensibilidad al contraste, perimetría, retinografía de campo amplio, autofluorescencia, tomografía de coherencia óptica (OCT), electrorretinografía (ERG), EOG y potenciales evocados, óptica adaptativa y pruebas genéticas.46,74-89
5.2.1.1. Pruebas subjetivas
Agudeza visual
La prueba de AV no confirma ni descarta el diagnóstico. Aun así, existe cierto consenso sobre la conveniencia de su medición al inicio del estudio diagnóstico y en cada visita. Esta medida, en algunas de las distrofias, puede conservarse hasta estadios muy tardíos y en otras puede ser el primer síntoma.18,70
muy baja
Sensibilidad al contraste
La evidencia identificada apunta a que es conveniente realizarla si se dispone de ella, ya que en algunas distrofias, como en la RP, los pacientes pueden beneficiarse de filtros de contraste para una mejor lectura. Sin embargo, esta prueba no parece aportar información sobre el diagnóstico.18,74
baja/muy
baja
Campos visuales
Es conveniente realizar una campimetría al inicio cuando existe la sospecha de distrofias extensas de la retina. Además, se debe tener en cuenta que en la RP esta medida se debe realizar por motivos legales en pacientes que todavía conservan CV. Dado que se produce una pérdida media del 4-5% anual no debería hacerse con una frecuencia superior a la anual.18, 75
baja/muy
baja
Test de colores
No se identificó evidencia que informe sobre la necesidad de realizar el test de colores a todo paciente con sospecha de DHR. No obstante, parece conveniente realizar esta prueba ante la sospecha de distrofia de conos y sospecha de acromatopsias.46,70
Adaptación a la oscuridad
No se identifican artículos que lo aconsejen de forma rutinaria.
baja/
moderada
5.2.1.2. Pruebas objetivas
Retinografía
No existen estudios con retinografía convencional, tan solo se describe el uso y la utilidad de las retinografías de campo amplio, especialmente en las distrofias extensas de la retina.76
muy baja
Autofluorescencia
Actualmente, es una de las pruebas más importantes para el diagnóstico de las distrofias de retina. Recomendada para el diagnóstico y seguimiento de distrofias tanto maculares77 como de retina periférica,18 aunque en distrofias maculares la autofluorescencia no siempre se correlaciona con la pérdida sensitiva.78
baja
Estudios observacionales en los que se realiza autofluorescencia de campo amplio apuntan a que las alteraciones en la autofluorescencia pueden preceder al daño en la retina.79,80 Asimismo, se ha valorado la utilización de diferentes tipos de autofluorescencia la realización de autofluorescencia de onda corta y de ondas cercanas al infrarrojo. Los datos apuntan a que siendo útiles ambas técnicas, la cercana al infrarrojo podría tener mayores ventajas para el estudio de la RP. Más de la mitad de los pacientes con RP presentan un anillo de hiper autofluorescencia perifoveolar.81
Se ha propuesto la autofluorescencia como prueba determinante en pacientes no colaboradores como en los niños, donde las pruebas electrofisiológicas son menos sencillas de realizar.73,82
Densitometría
No existen publicaciones que avalen su uso sistemático.
muy baja
AFG
En enfermedades como la RP es recomendable la realización de una AFG en caso de aparición de complicaciones como el edema macular. En la enfermedad de Stargardt clásicamente se describe el silencio coroideo de la AFG que es caso patognomónico de esta enfermedad, aunque con la aparición de la autofluorescencia y la OCT parece perder importancia en el diagnóstico inicial.18
muy baja
OCT
La OCT es una prueba imprescindible para el diagnóstico de posibles complicaciones relacionadas con la RP, ya que hasta un 38% de los pacientes pueden cursar con edema macular cistoide.18,83
El estudio de la retina externa con OCT de dominio espectral (concretamente la línea de los elipsoide) se relaciona con la funcionalidad de la retina en pacientes con RP.81
La OCT se considera como el gold standard para el diagnóstico de la retinosquisis congénita ligada al cromosoma X, incluso en niños muy pequeños donde no es posible realizar otras pruebas la OCT nos facilita el diagnóstico.72
La OCT también puede ayudar en el diagnóstico de las distrofias maculares.77
baja/muy
baja
Pruebas electrofisiológicas
En la RP y en otras enfermedades extensas de la retina es recomendable realizar un ERG al inicio y si es posible un ERG en patrón. Existen enfermedades hereditarias que típicamente nos darán ERG negativo. Una vez que se recoge un ERG plano no vale la pena repetirlo. Un ERG de patrón plano (ERG-P) indica alta probabilidad de pérdida visual severa en menos de cinco años. El ERG multifocal (ERG-mf) puede servir para monitorizar la función central.18
baja/muy
baja
La realización de ERG y de ERG-P es aconsejable cuando exista sospecha de distrofia macular,18,84 aunque el ERG convencional puede ser normal o estar solo levemente alterado en muchas de las distrofia maculares, como la enfermedad de Stargardt, Best y distrofias viteliformes de la edad adulta.84
El ERG fotópico alterado con escotópico conservado puede ser útil para el diagnóstico de las distrofias de conos.85 El ERG-mf también puede ser útil para monitorizar distrofias maculares. El EOG es casi imprescindible para el diagnóstico de la enfermedad de Best, en estos pacientes se aprecia un EOG alterado con un índice de Arden patológico y normalidad en el ERG.18
La inclusión del ERG y el potencial evocado visual (PEV) es útil en el protocolo de exploración clínico en la sospecha de un cuadro sugestivo de ACL.26
Se ha descrito el uso de PEV específicos por colores en pacientes con distrofias de conos, aunque no parece que sea una técnica estandarizada.86
Especial interés toman estas pruebas cuando se busca poder detectar precozmente una DHR sindrómica, dado que existen múltiples síndromes multiorgánicos en que aparecen signos y síntomas oculares similares a los que se observan en los casos aislados de DHR ya que la afectación del PVE con ERG normal sugiere una afectación de la vía visual y no de la retina90,91 aun cuando, en ocasiones, los hallazgos oftalmológicos pueden ser sutiles y/o aparecer a posteriori.92
muy baja
Óptica adaptativa
Hasta la fecha se han realizado pocos estudios con óptica adaptativa en distrofias de retina, pese a que no confirman el diagnóstico se ha demostrado que en la RP se encuentran alteraciones maculares por óptica adaptativa incluso en pacientes con una buena función visual central.87,88
muy baja
Prueba genética
El diagnóstico genético supone a día de hoy un reto, dada la enorme heterogeneidad de las alteraciones genéticas que podemos encontrar.89
Sin embargo, gracias a la incorporación de las técnicas de secuenciación masiva (NGS, del inglés: next generation sequencing) es factible caracterizar genéticamente más del 50% de los casos, en un plazo rápido y a un coste asequible.70-72
Actualmente el diagnóstico genético es importante para confirmar o refinar el diagnóstico clínico, especialmente en el estudio de casos sindrómicos o casos poco claros. Es recomendable también en la enfermedad de Stargardt, para el asesoramiento genético, la planificación familiar, en casos de sospecha de mujeres portadoras de enfermedades XL y será importante en un futuro para posibles tratamientos individualizados.18,89 Para más información ver 5.2.5. Estudio genético
baja/muy
baja
Otras pruebas
Otras pruebas pueden ser de utilidad ante la sospecha de algunas enfermedades tales como la ACL.26 Así, en la sospecha de un cuadro sugestivo de ACL se recomienda el protocolo de exploración clínico siguiente:26
Dentro de la exploración obligatoria, además de la evaluación oftalmológica y de la realización de un ERG y PEV, ante la sospecha de una forma sindrómica de la enfermedad se debe realizar:
- Evaluación neurológica y sistémica
- Resonancia magnética nuclear cerebral
- Examen ecográfico de abdomen y pelvis (renal)
- Examen radiológico de las manos
- Evaluación de la función hepática y renal
Además, a veces es recomendable realizar las siguientes exploraciones:
- Exploración metabólica enzimática (ácidos grasos de cadena larga, lactato y piruvato séricos, ácidos orgánicos urinarios, aminoacidemia y aminoaciduria, ácido fitánico, sialotransferrina)
- Enzimas musculares
- Potenciales evocados auditivos de tronco cerebral
baja/muy
baja
Resumen de la evidencia
| Calidad muy baja | La evaluación de la AV es la primera prueba a realizar cuando existe una sospecha de DHR.18,70 |
| Calidad baja / muy baja | Ante una sospecha de DHR, es adecuada la realización de un estudio del CV, al menos al inicio. Dada la evolución del CV en pacientes con DHR, no aporta demasiada información su evaluación con periodicidad inferior al año.2,56 |
| Calidad baja | La autofluorescencia se confirma como prueba imprescindible tanto en distrofias extensas como en distrofias maculares. Esta técnica puede ser de gran utilidad en niños.18,77,79-82 |
| Calidad baja/muy baja | La OCT es una prueba imprescindible en el diagnóstico de las distrofia maculares,77 es el gold standard para el diagnóstico de la retinosquisis congénita ligada al cromosoma X72 y se ha demostrado útil en el diagnóstico y control de las complicaciones asociadas a las distrofias extensas de retina como el edema macular en la RP.18,81,83 |
| Calidad baja/muy baja | Las pruebas electrofisiológicas siguen siendo imprescindibles en el diagnóstico de las distrofias de retina. Es aconsejable la realización de ERG cuando exista sospecha tanto de distrofias centrales como periféricas.18,84 |
| Calidad baja | Es aconsejable la realización de ERG cuando exista sospecha de DHR sindrómica,90,91 dado que los hallazgos oftalmológicos pueden ser sutiles y/o aparecer a posteriori.92 |
| Calidad muy baja |
El electrooculograma asociado a ERG puede ser definitivo en el diagnóstico de la enfermedad de Best.18 |
| Calidad baja / muy baja | Actualmente en niños, casos dudosos, casos sindrómicos, pacientes con enfermedad de Stargardt, mujeres en riesgo o diagnóstico confirmado de ser portadoras de RP ligada al cromosoma X y para la planificación familiar es conveniente valorar el diagnóstico genético.18,89 |
| Calidad baja / muy baja | En un futuro el diagnóstico genético será importante para posibles tratamientos individualizados.18,89 El diagnóstico genético permite confirmar el diagnóstico e identificar portadores con riesgo de transmitir la enfermedad.89 |
| Calidad muy baja |
El uso de retinografías de campo amplio puede ser útil en la monitorización de las distrofias extensas de retina.76 |
| Calidad muy baja | La AFG, que clásicamente se había utilizado para el diagnóstico de posibles complicaciones, ha perdido interés tras la aparición de sistemas no invasivos como la OCT.18 Pero sigue siendo útil en la detección del silencio coroideo casi patognomónico de la enfermedad de Stargardt (sólo se describe también en la intoxicación por plata). |
| Calidad muy baja | El ERG-mf y el ERG-P pueden ayudar en la monitorización funcional del área macular de los pacientes afectos de DHR.76 |
| Calidad muy baja | Los test de colores y la sensibilidad al contraste pueden estar indicados ante determinadas sospechas como la existencia de una distrofia de conos.18 |
| Calidad muy baja | Otras pruebas pueden ser de utilidad ante la sospecha de ACL.26 Así, en la sospecha de un cuadro sugestivo de ACL asociada a enfermedad sistémica se recomienda, como mínimo, el protocolo de exploración clínico siguiente:26
|
| No existe evidencia disponible que aconseje la utilización del test de adaptación a la oscuridad, aunque estos pueden ayudar a distinguir una ceguera nocturna patológica de la que puede ser habitual en pacientes miopes. | |
| No existen estudios disponibles que avalen el uso sistemático de la densitometría retiniana. | |
| No existe evidencia suficiente que avale el uso de técnicas de óptica adaptativa en pacientes con distrofia de retina, aunque puede ser una herramienta interesante para un futuro. |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- Calidad global de la evidencia: la evidencia deriva de estudios de cohortes y revisiones de series de casos, considerándose la calidad global de la evidencia como muy baja.
- Balance entre beneficios y riesgos: Se considera que el potencial beneficio de la realización de las pruebas indicadas para el diagnóstico o confirmación del diagnóstico supera los riesgos de las mismas.
- Valores y preferencia de los pacientes: No se identificaron estudios publicados sobre valores y preferencias de las personas con DHR en cuanto a esta pregunta. En el estudio cualitativo realizado en el contexto español, las pruebas cuentan con una valoración positiva por parte de los pacientes especialmente si tienen una finalidad diagnóstica.
- Aceptabilidad: En el estudio cualitativo con pacientes con DHR en el contexto español, los pacientes perciben que las pruebas no están exentas de molestias específicas para las personas con DHR, o barreras por lo que resaltan los siguientes aspectos:
- Que se facilite el acceso a las pruebas (por ejemplo, evitar el uso de pantallas con número para pasar a un servicio, ya que éstas no están adaptadas a los pacientes con baja visión o sin visión puesto que no son parlantes).
- Que se adapte el lenguaje usado (por ejemplos, recibir referencias espaciales verbalmente específicas -a su derecha, delante, detrás…-, en lugar de gestuales).
- Que el profesional sanitario acompañe a los pacientes en los traslados entre consultas y acceso a las pruebas, especialmente si la persona no va acompañada por un familiar o conocido.
- Que se realicen pruebas adaptadas a las condiciones específicas de baja visión de cada patología y a su resto visual.
- Factibilidad: Estas técnicas no suelen estar disponibles en todos los hospitales.
El grupo elaborador formuló las siguientes recomendaciones considerando las opciones que podrían ayudar al diagnóstico y confirmación de las DHR.
Recomendaciones
| Fuerte | Ante la sospecha de DHR, se recomienda que se realice una exploración oftalmológica completa y dirigida (medidas de AV, CV, FO, pruebas electrofisiológicas, test de colores, autofluorescencia y OCT). |
| Condicional | En niños pequeños y pacientes no colaboradores se sugiere el uso de la electrofisiología, la autofluorescencia y de la OCT. |
| Condicional | Ante la sospecha de cualquier DHR sindrómica en ausencia de signos oftalmoscópicos evidentes en la exploración, se sugiere el test de electrofisiología para su diagnóstico. |
5.2.2. Estudio electrofisiológico
- ¿Cómo debe hacerse el estudio electrofisiológico? ¿A quién hay que pedírselo? ¿qué pruebas hay que solicitar?
Para el desarrollo de esta pregunta se ha tomado como referencia los estándares o indicaciones que la Sociedad Internacional de Electrofisiología Clínica y Visión (International Society for Clinical Electrophysiology of Vision-ISCEV) ha establecido para la realización de todas y cada una de las pruebas electrofisiológicas, de tal manera que los resultados sean interpretables por otra persona, reproducibles y comparables con otros que sean realizados por otro equipo profesional.93-97 En la que se muestra a continuación, se recogen de manera resumida.
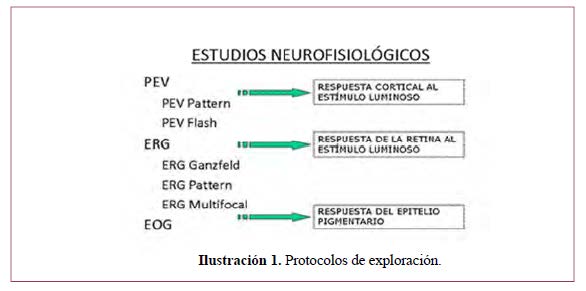
A. Potencial evocado visual (PEV)94
Muchas son las herramientas diagnósticas que permiten estudiar el ojo tanto en el segmento anterior como en el posterior, sin embargo, son necesarias otro tipo de pruebas si queremos estudiar las estructuras neurológicas que sirven de puente entre el ojo y el cerebro. Para ello, se dispone de los PEV, que no son más que la medida de la respuesta eléctrica de la corteza cerebral occipital ante estímulos luminosos, ya que «vemos con el cerebro».
Desde que un estímulo visual llega al ojo hasta que el cerebro lo procesa pasa un determinado tiempo, del orden de milisegundos. Las variaciones en ese tiempo o en la forma de la respuesta que se obtiene informan si hay un problema en la vía de comunicación nerviosa entre el ojo y el cerebro. La ISCEV dispone de unos protocolos consensuados y revisados cada cuatro años por científicos pertenecientes a esta sociedad.94
Para medir esas variaciones son precisos electrodos sobre distintos puntos de la cabeza, donde se puede registrar la actividad eléctrica. Es necesario usar pastas abrasivas y conductoras para conseguir una menor resistencia al paso de la corriente eléctrica, limitada por la piel y cualquier producto químico aplicado en la zona, como sucede con lacas y gominas.
El paciente debe estar sentado y relajado, para evitar registrar actividad eléctrica muscular o variaciones de la señal por movimientos. En el caso de pacientes que precisen refracción, deberán usarlas para realizar la prueba con la mejor corrección.
Se hacen dos tipos de pruebas en clínica, dependiendo del estímulo:
- PEV con estímulo Flash o haz de luz difusa
- PEV con estímulo Pattern o damero reversible (PEVP)
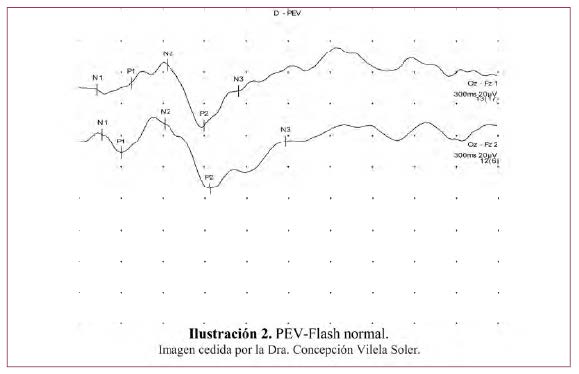
A.1. Protocolo clínico del PEV con estímulo Flash
El PEV como respuesta a un Flash, que son destellos de luz blanca intermitentes, no necesita la fijación, valorando solo el paso de la señal luminosa hasta la corteza cerebral. Se obtiene una gráfica con cinco puntos N1, P1, N2, P2 y N3. Se valoran variaciones de latencia de la N2. En la Ilustración 2 puede verse una respuesta normal al PEV-Flash con sus cinco componentes.
Aplicación en clínica: en los casos de imposibilidad de correcta fijación, en niños pequeños que no fijan (por debajo de tres meses), malos colaboradores, o en caso de coma. Se puede hacer con el estimulador Ganzfeld o en pacientes encamados, con las gafas de leds goggles.98,99 Otra utilidad clínica es para aquellos pacientes con opacidades de medios que impiden valorar FO (por ejemplo una catarata o una hemorragia vítrea), ya que si se recoge respuesta en esta prueba, puede pensarse en realizar cirugías que traten de resolver esas opacidades, algo que se descartará si el PVE con flash es plano.
Esta prueba es tan solo la confirmación del paso de señal luminosa de por la vía visual, no implica la función sensorial de «ver» correctamente.
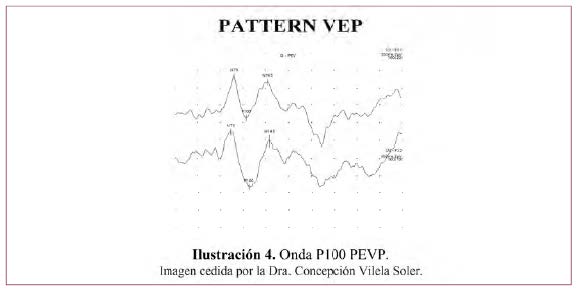
Para la obtención del PEVP es necesaria la fijación de la visión del paciente en un punto y solo aparecerán respuestas corticales si la fijación es correcta. Da información del paso del estímulo y del tiempo que tarda en pasar. Se obtiene una gráfica con tres puntos de inflexión N75, P100 y N145, que indican la polaridad N=negativo, P=positivo y con el número, el tiempo en milisegundos que tarda en aparecer dicho punto. Se consideran variaciones de la onda P100 (Ilustración 4) que se expresa en milisegundos y microvoltios, comparados con patrones normales del propio laboratorio.
No se utiliza mucho en clínica humana por ser muy poco sensible y por su enorme variabilidad interindividual, que hace que sólo sea útil en la valoración de la simetría de la respuesta entre ambos ojos, así como en constatar que se mantiene el registro de una respuesta en ambos ojos (la latencia de las ondas positivas debería de ser inferior a 150 milisegundos).
A.2. Protocolo clínico del PEV con estímulo Pattern (PEVP)
Pupilas: no alteradas por drogas midriáticas o mióticas. Conseguir la mejor AV. Se debe hacer estimulación monocular. Estimulación binocular solo cuando hay poca colaboración. Posición relajada para minimizar el artefacto de músculos. Si no consigue fijación: Aumentar ángulo/punto de fijación. Si sigue sin fijar: «Fijación al centro» o pasar a PEV-Flash.
Estimulación: Damero que revierte (pattern reversal)
Ilustración 3: cuadros blancos y negros o barras que cambian de negro a blanco y de blanco a negro continuamente. El estímulo se definirá en términos de ángulo visual de cada cuadro, expresado en grados. Se deben usar por indicación de ISCEV dos medidas por lo menos: 15 minutos (cuadro pequeño) y 60 minutos (cuadro grande). Se realiza siempre con el paciente sentado a un metro de distancia. Es absolutamente necesario que el paciente porte su corrección, pues los defectos de refracción impiden ver el damero y alteran falsamente la respuesta a este tipo de estímulo. Hay que estimular cada ojo por separado.

A.3. Aplicaciones clínicas del PEV
Enfermedades del nervio óptico: neuropatía óptica isquémica, desmielinizante, traumática, por tumores, inflamatoria, por tóxicos (alcohol, tabaco, metotrexato, etc.). También se afecta en alteraciones funcionales como la ambliopía o en opacidades de medios, pues estos impiden la correcta visualización del estímulo, y también en el glaucoma, y las maculopatías.98,100,101 En todos estos casos permitirá confirmar un diagnóstico que se sospechaba, realizar un diagnóstico que no se sospechaba, evaluar discrepancias entre signos y síntomas (simuladores), monitorizar la progresión de la enfermedad, descartar algunos diagnósticos, etc.
Esta prueba permite (sola o junto con el ERG-P) diferenciar enfermedades maculares de neuropatías ópticas. También permite diferenciar neuropatías ópticas desmielinizantes que retrasarán la latencia de la P100, de aquellas que cursan con disminución de fibras de las células ganglionares como suceden en las neuropatías ópticas isquémicas o en muchas de las hereditarias, pues estas últimas provocarán fundamentalmente una reducción en la amplitud de la onda P100.
B. Electrorretinograma (ERG)
El ERG es el registro de los cambios de potencial eléctrico obtenido en la retina tras un estímulo luminoso. La ISCEV facilita unos protocolos de cada tipo de ERG que actualiza cada cuatro años.93,95,97
Existen diferentes tipos de ERG según el estimulador:
- ERG de campo lleno (Ganzfeld), o campo completo
- ERG con damero alternante (Pattern)
- ERG-mf
B.1. ERG-Ganzfeld (ERG-G)93
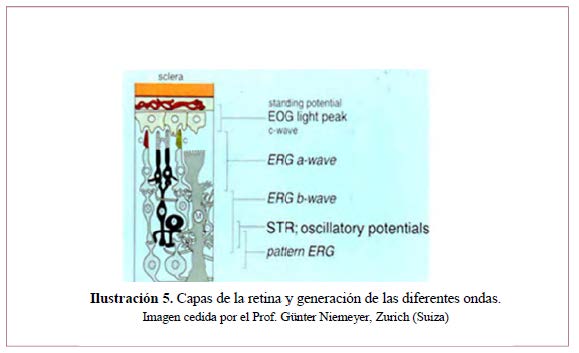
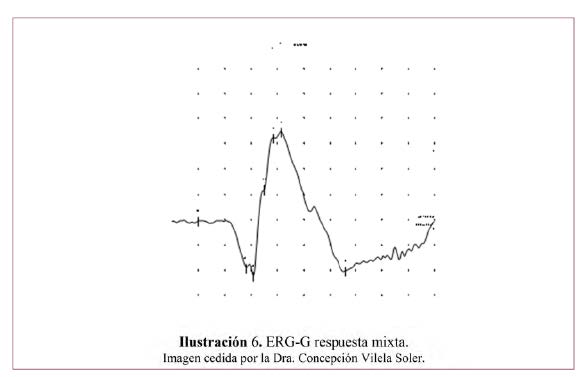
Es una respuesta en masa evocada en toda la retina por un destello de luz breve.93 Se manifiesta como una onda cuyos principales componentes representan fundamentalmente la respuesta de fotorreceptores: onda a negativa (por la hiperpolarización de los fotorreceptores debido a la salida de Na+ tras el estímulo luminoso) que se origina en los fotorreceptores, seguida de una onda b positiva (por la despolarización de las células de la retina interna debido a la entrada de K+ en su interior) que procede principalmente de la actividad de células bipolares y de Müller. Sobre la rama ascendente de la onda b veremos unos pequeños picos de onda, los potenciales oscilatorios reflejando la actividad a nivel de la capa plexiforme interna. En la ¡Error! No se encuentra el origen de la referencia. podemos observar las diferentes capas de la retina y su función en la generación de las ondas que componen el ERG. En la Ilustración 6 vemos el ejemplo de una respuesta mixta (conos y bastones) en condiciones escotópicas.
La retina convierte la energía fotópica en señales eléctricas y procesa estas señales en patrones complejos que son transferidos al cuerpo geniculado externo y de ahí al córtex visual del lóbulo occipital. Esta vía nerviosa está constituida por los fotorreceptores y tres neuronas.
En la capa plexiforme externa, los axones de los conos y los bastones establecen sinapsis con las dendritas de la 1ª neurona –la célula bipolar y también con las células horizontales. La célula bipolar (núcleo, capa nuclear interna) sinapta en la plexiforme interna con la 2ª neurona – la célula ganglionar y también con las células amacrinas. Los axones de las ganglionares forman la capa de fibras nerviosas que a través del nervio óptico llegan al cuerpo geniculado externo donde hacen sinapsis con la 3ª neurona cuyos axones a través de radiaciones ópticas llegan al córtex occipital.
Características de los componentes del ERG-G
En la descripción de las distintas ondas del ERG nos tenemos que fijar en una serie de características:
- Morfología (Ilustración 6)
- Amplitud de las ondas:
- onda a (desde la línea de base hasta el valle)
- onda b (desde el valle de la onda a hasta el pico de la onda b
- Características temporales:
- la latencia (tiempo desde el estímulo al inicio de la onda)
- tiempo de culminación (tiempo desde el estímulo hasta el pico de la onda)
- Parámetros de las ondas:
- Duración de la respuesta 150 ms.
- Latencia onda a 12-15 ms y de la onda b 40-45 ms.
- Amplitud de las ondas a y b.
- Cociente b/a (normal >2).
Protocolo clínico102
Condiciones del paciente:
- Dilatación pupilar con colirio de tropicamida.
- Pre-adaptación a la luz y la oscuridad:
- 20 min de adaptación a la oscuridad para el registro de la respuesta de bastones y mixta.
- 5-10 min de adaptación a la luz para la respuesta de conos.
- Fijación: se debe incorporar un punto de fijación a la campana de estímulo. Un ojo estable es importante para que los movimientos del ojo no artefacten y no bloqueen la luz al parpadear.
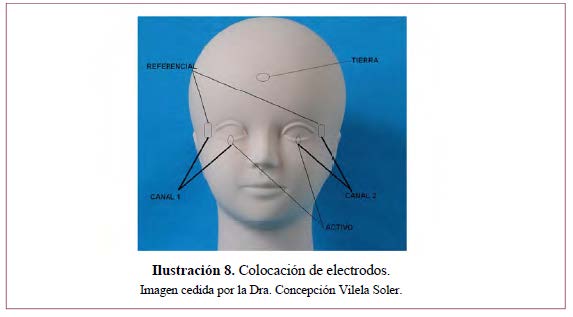
Técnicas para la recogida (Ilustración 7 e Ilustración 8):
- Se recomienda un estimulador de campo amplio (Ganzfeld).
- Electrodos activos o de recogida: lentes de contacto corneal, hilos y fibras conductores de contacto conjuntival103.
- Electrodos de referencia: reborde orbitario ipsilateral.
- Electrodo de tierra: localización típica frontal central.

Condiciones del registro:
- Tiempo de análisis: 150-200 ms. A ser posible con un retardo de 25 ms para ver la línea de base.
- Frecuencia de estimulación:
- Luz azul escotópica: Intervalo mínimo de dos segundos entre destellos (0,5 pulsos por segundo).
- Luz blanca escotópica: Intervalo mínimo de 10 segundos (0,1 pulsos por segundo).
- Luz blanca fotópica: Intervalo 0,5 segundos (2 pulsos por segundo).
- Potenciales oscilatorios: Intervalo 1,5 segundos en ojos adaptados a la luz y 15 segundos en ojos adaptados a la oscuridad.
- Flicker: 30 estímulos por segundo.
- Número de estímulos: De uno a cuatro. Se recomiendan dos series.
- Filtros: Las normas internacionales aconsejan banda pasante entre 0,3 Hz y 300 Hz con posibilidad de ampliar a 30 Hz y 1000 Hz para obtener los potenciales oscilatorios.
Respuestas específicas
Hay cinco respuestas básicas estandarizadas por la ISCEV93:
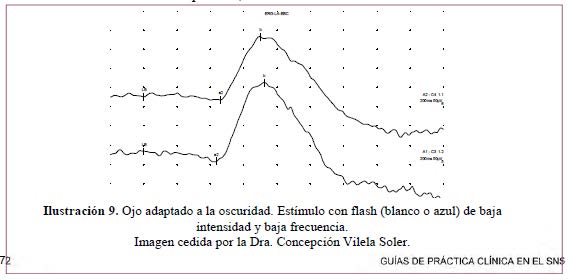
-Respuesta de bastones– Ilustración 9
- Ojo adaptado a oscuridad 20 minutos.
- Estímulo: destello blanco o azul tenue de una potencia de 2,5 unidades logarítmicas por debajo del flash estándar, definido como el que produce un estímulo de 3,43 cd/m2/seg expresado en energía luminosa por m2) en la superficie de la campana Ganzfeld.
- El resultado es una deflexión positiva, onda b.
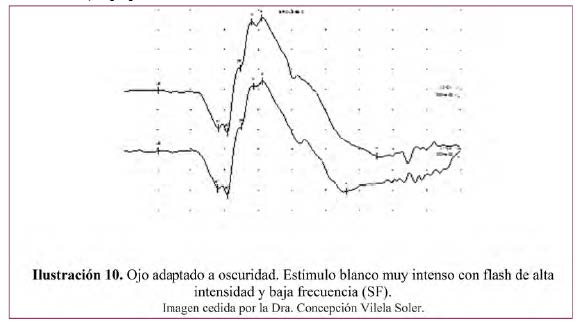
– Respuesta máxima– Ilustración 10.
- Respuesta mixta conos y bastones en ojo adaptado a la oscuridad, producida con un estímulo blanco muy intenso (SF) de candela por metro cuadrado.
- Genera amplitudes grandes de las ondas a (fotorreceptores) y b (células bipolares y de Müller) y Potenciales oscilatorios con origen en las capas media de la retina (células amacrinas) superpuestos en la onda b ascendente.
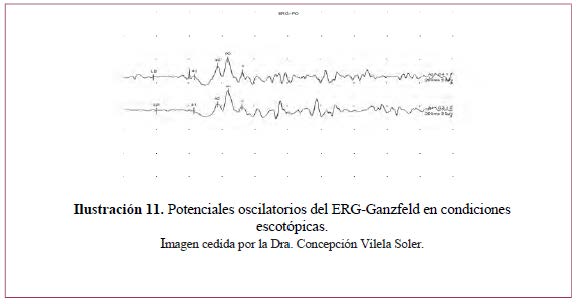
– Potenciales oscilatorios– Ilustración 11.
- Se pueden obtener con ojo adaptado a la luz o a la oscuridad, con un estímulo blanco intenso (SF).
- Se originan en la capa media de la retina, (células amacrinas), muy sensibles a la hipoxia.
- Se obtiene de forma aislada, ampliando la banda pasante para filtrar los componentes más lentos del ERG. Valoraremos cuatro ondas.
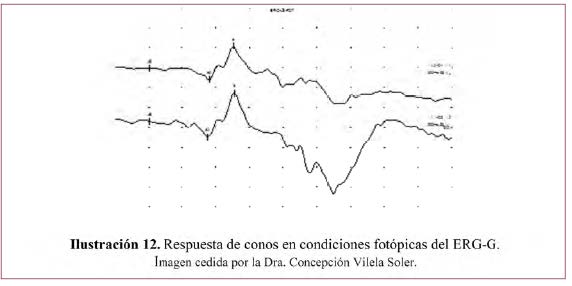
– Respuesta de conos– Ilustración 12.
Respuesta de conos a estímulo único
- Ojo adaptado a la luz 5-10 minutos.
- Iluminación de fondo de 17 a 34 cd/m2 que bloquea los bastones.
- Se estimula con un destello blanco (SF) único que no debe repetirse a intervalos menores de 0,5 segundos.</li<
- Se puede promediar para mejorar la relación señal/ruido.
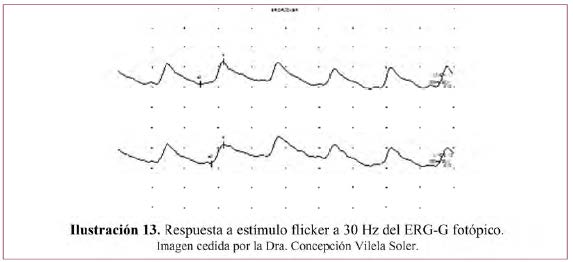
– Respuesta de Flicker– Ilustración 13.
- Se obtiene con un estímulo máximo bajo la misma iluminación de fondo supresora de bastones.
- Los destellos se presentan con una frecuencia de 30 Hz que permiten aislar todavía más la respuesta de los conos.
- Se registran cuando se han estabilizado las ondas.
Interpretación del ERG-G
El ERG de campo completo permite diferenciar si son fundamentalmente los conos o los bastones los que están alterados. Ello se basa en:
- Diferenciar conos/bastones por la sensibilidad de los fotopigmentos a distintos estímulos luminosos:
- Bastones (Rhodopsina) max 504nm-azul condic escotópicas
- Conos: sensibilidad max 555nm-verde-amarillo fotópico
- También se les diferencias por sus distintos tiempos refractarios:
- Las respuestas de los conos se fusionan si la frecuencia de un estímulo fluctuante es de 50-60Hz y la de bastones si es 20Hz, por lo tanto si usamos el flicker a 30Hz obtendremos una respuesta básicamente pura de conos
- Además:
- La respuesta de conos será de baja amplitud y latencia corta
- La de bastones será de gran amplitud y latencia prolongada
Por su parte, también podremos diferenciar si la afectada es la retina interna o la externa, así la alteración de la onda a implicará una disfunción de la retina externa (fotorreceptores), mientras que en las disfunciones de la retina interna se alterará el registro de la onda b y desaparecerán los potenciales oscilatorios.
Patrones de afectación:
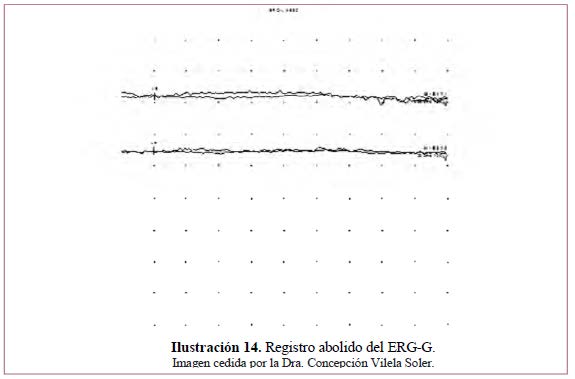
• Registro abolido: traduce la afectación de la retina en toda su extensión con afectación de fotorreceptores y células bipolares. Ilustración 14.
Enfermedades en que se encuentra este patrón: RP y coroideremia en estadios avanzados, desprendimientos de retina totales, retinopatía asociada al carcinoma, enfermedad de Goldman- Fabre.
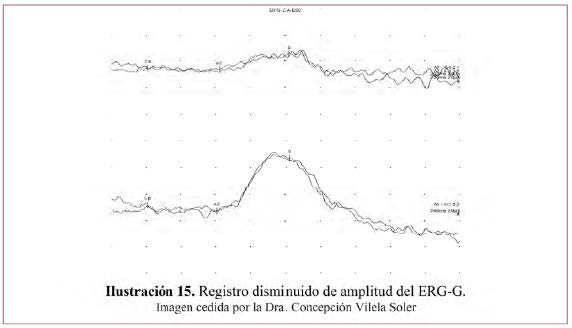
• Registro disminuido: la amplitud de la ondas a y b está disminuida. Ilustración 15.
Enfermedades en que se encuentra este patrón: enfermedades extensas de la retina externa y la coroides, formas incipientes de RP, desprendimiento de retina parcial.
• Registro negativo – Ilustración 16.
«ERG decapitado o negativo»: onda b disminuida con onda a normal. Cociente b/a <2
Informa de una afectación selectiva de las células bipolares o de Müller con actividad normal del fotorreceptor.
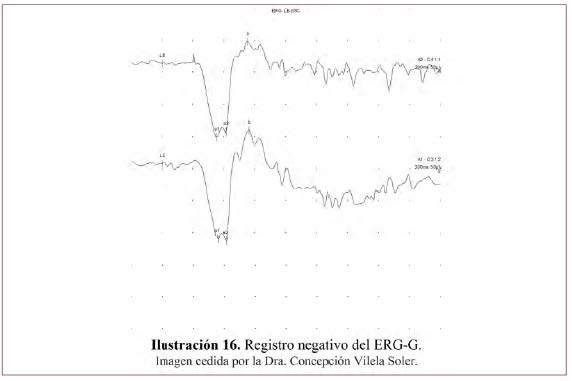
Enfermedades en que se encuentra este patrón (posibles causas de «ERG decapitado o negativo»):6
- Patología vascular de la arteria y vena central de la retina.
- Retinosquisis ligada al X.
- En la ceguera nocturna congénita estacionaria tipo Schubert–Bornschein de herencia AR o recesiva XL cursa con ausencia completa de onda b pero con onda a preservada, la amplitud de esa onda a aumenta al aumentar la intensidad del estímulo luminoso sin hacerlo la onda b. Puede cursar con FO normal (las más frecuentes) o anormal (Oguchi’s / fundus albipunctatus / retina parcheada de Kandori). Esa ausencia es más evidente en los registros escotópicos.
- Toxicidad por metanol o quinina.
- Enfermedades por acúmulo de lipopigmentos.
- Retinopatía asociada al melanoma.
• REGISTRO ESCOTÓPICO ANORMAL CON FOTÓPICO NORMAL.6
Enfermedades en que se encuentra este patrón: Registro típico de la forma más frecuente de inicio de la RP (Ilustración 17), ceguera nocturna congénita.

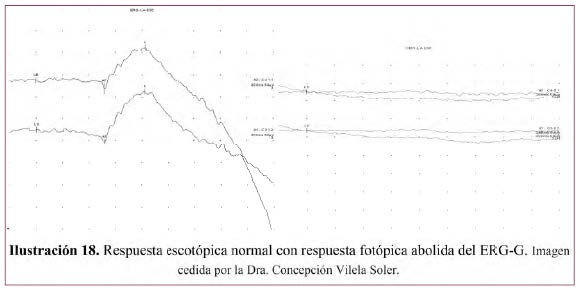
• REGISTRO ESCOTÓPICO NORMAL CON FOTÓPICO ANORMAL (Ilustración 18).6
• Enfermedades en que se encuentra este patrón: Distrofia de conos, monocromatismo de bastones también llamado acromatopsia congénita, Monocromatismo de conos azules ligado a X
Factores que influyen en la respuesta del ERG-G:
- La intensidad del estímulo y el tamaño de la pupila.
- El ERG-G es poco sensible a los errores de refracción, excepto en ojos con miopía magna que pueden tener respuestas disminuidas.
- Los pacientes ancianos pueden mostrar respuestas de amplitud disminuida.
- Los recién nacidos tienen una señal ERG pequeña pero aumenta rápidamente en los primeros meses de vida.
A los niños se les puede colocar electrodos de contacto en casos de sedación. Según Kriss et al.104 del Great Ormond Street Hospital, para una rutina de exploraciones, se utilizan los electrodos de registro infraorbitarios y se valora la amplitud de la respuesta que suele ser un 50% menor.
Aplicaciones clínicas del ERG-G
Diagnóstico de confirmación y evolución en enfermedades hereditarias de la retina. También para identificar a pacientes portadores de esas enfermedades sin síntomas clínicos o anormalidades en el FO. Estas incluyen:
- RP y coroideremia, también para identificar a pacientes portadores sin síntomas clínicos o anormalidades en el FO.105-107
- Ceguera nocturna estacionaria congénita.
- Disfunción de conos. Distrofia de conos y distrofia de conos y bastones, acromatopsia congénita.
- Detección precoz de retinopatías tóxicas (vitaminas o fármacos)
- Evaluar la función retiniana en déficit de vitamina A, gran reducción de amplitud.101
- Evaluar presencia o ausencia de actividad retiniana.
- Estudio de la pérdida de AV inexplicable.
- Estudio de pacientes con enfermedad de Parkinson.105,108,109
B.2. ERG-pattern (ERG-P)97
Es un potencial retiniano generado en las células ganglionares de la retina evocado por un estimulador de damero o barras alternantes. La ISCEV ofrece un protocolo de realización, actualizado cada cuatro años.97
Puede darnos información de la actividad de las células de la retina interna y de la mácula de forma selectiva y puede aparecer alterado complementando el resultado de un ERG-G normal.
Protocolo clínico
El registro del ERG-P es de una amplitud muy baja, por lo que a veces se altera sin que necesariamente haya patología ocular. Por ejemplo, necesita colaboración del paciente, puede alterarse por ejemplo si el paciente tiene actividad cerebral intensa (si está pensando en algo que le altera, las ondas beta cerebrales pueden ocultarla) o si hay actividad muscular cercana a los electrodos (ej.: si hace fuerza con la mandíbula y el masetero tiene importante actividad eléctrica). Por eso hay que pedir al paciente que deje la mente en blanco y que no cierre la boca apretando la mandíbula.
Condiciones del paciente:
- Posición: lo más cómoda posible, relajado y con la mente en blanco. Recordar al paciente que no debe contraer el cuello ni apretar la mandíbula ya que siendo una respuesta pequeña se puede alterar por el ruido muscular.
- Pupilas: sin dilatación.
- Refracción: los pacientes deben estar con la corrección óptica adecuada.
- Fijación: un punto de fijación en el centro de la pantalla.
- Se hacen series monoculares y binoculares
Técnica de recogida:
- Electrodos activos de contacto corneal o próximos a la conjuntiva bulbar, colocados en el punto medio del párpado inferior para que no interfieran la visión.
- Electrodos de referencia: se deben colocar en el canto externo del ojo.
- Electrodo de tierra: la frente es la localización típica.
Parámetros de estímulo:
- Para el ERG-P básico se necesita un damero alternante de cuadros blancos y negros con unas medidas de campo entre 10 y 16º, y un tamaño de cuadro de 0,8º, con un contraste lo más próximo al 100% y no menor del 80% y una luminosidad fotópica de 80 cd/m2.
- Frecuencia del estímulo: de 1-3 Hz para el ERG-P transitorio. El repetitivo (steady-state) ERG-P se obtiene con frecuencia de 8Hz y evoca una onda final sinusoidal que requiere un análisis de Fourier para su valoración.
- Número de estímulos: Siempre es necesario hacer promediación de 150 estímulos o más por el tamaño tan pequeño de la señal.
- Filtros: banda pasante de 1 a 100Hz.
Respuesta del ERG-P
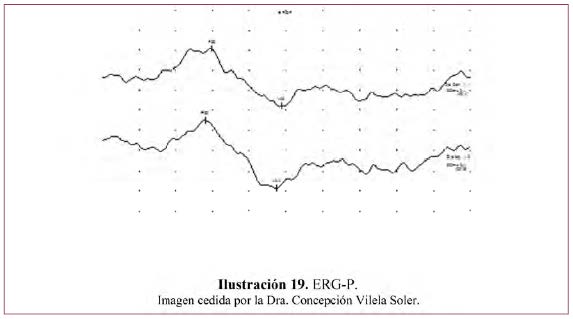
- La morfología de la respuesta del ERG-P transitorio es una onda negativa a 35 ms (N35), una onda positiva a 50 ms (P50) seguida de un componente alargado a los 95 ms (N95) (Ilustración 19).
- Se valora la amplitud interpicos de la P50 y N95 que no debe ser menor de 5 microvoltios y la latencia de la P50.
- Los parámetros de normalidad deben establecerse en cada laboratorio, sin olvidar que el ERG-P cambia con la edad.
Aplicaciones clínicas del ERG-P:
El ERG-P estará siempre alterado en pacientes con defectos de refracción no corregidos, opacidades de medios y ambliopía, ya que al igual que el PVE en patrón reverso, se necesita que el paciente visualice el estímulo. Además se encontrará afectado en:
- Maculopatías, en esta patología suele estar siempre alterado y en ocasiones abolido. En la enfermedad de Stargardt, se pueden encontrar tres grupos dependiendo de la relación del ERG-P y el ERG-G. La alteración aparece siempre en la onda P50 que da información de la función macular.110
- En retinopatías extensas con ERG-G abolido; el ERG-P puede ser de utilidad en la monitorización y detección precoz de la afectación macular.111
- Enfermedades de nervio óptico: que afecta a las células ganglionares cuyos axones conforman el nervio óptico. En este caso la afectación aparecerá en la onda N95 lo que dará información directa de la función de las células ganglionares.110
- Neuritis óptica desmielinizante o isquémica
- Neuromielitis óptica sin esclerosis múltiple100
- Patología compresiva del nervio y quiasma óptico
- Enfermedades primarias hereditarias de las células ganglionares:
- Neuropatía óptica de Leber
- Atrofia óptica dominante
En general se puede utilizar el siguiente esquema para diferenciar enfermedad macular de enfermedad del nervio óptico:
| Maculopatía ERG-P alterado siempre Amplitud P50 disminuida o abolida Latencia P50 aumentada en ocasiones N95/P50 normal (>0,1) |
Enfermedad desmielinizante n. óptico ERG-P alterado en un 40%100,110 La latencia de la N95 alterada en el 85%100,110 La P50 abolida sólo en enf grave N95/P50 puede estar disminuída |
B.3. ERG-mf95
Consiste en la estimulación simultánea de diferentes áreas de la retina central, permitiendo la obtención de un mapa topográfico de la respuesta de los 30 a 50º centrales de la retina. La ISCEV ofrece un protocolo para la realización de esta prueba.95
Se utiliza en clínica para detectar defectos focales. No debe reemplazar al ERG-G, que es la exploración de elección, cuando se sospecha afectación generalizada de la retina o del sistema de bastones.
Técnica de recogida:
- Electrodo activo de contacto corneal o próximo a la conjuntiva bulbar que no interfiera en la visión y referencial en el reborde orbitario ipsilateral. Tierra en Fz. Se puede usar diferentes tipos de electrodo, HK-Loop, M.Hawlina112 o ERG-Jet o Gold-Foil
- El registro se realiza con dilatación pupilar
- Se hace en condiciones fotópicas
- El paciente se sitúa a 30 cm frente al monitor con la vista fija en un punto en el centro del mismo
- Se coloca la refracción adecuada
- Precisa la máxima colaboración del paciente manteniendo la fijación central durante toda la prueba
- El registro puede ser binocular o monocular. La ISCEV recomienda hacer un registro monocular para mejor información95
- Condiciones del registro:
- El estímulo se presenta en una pantalla de luminancia constante divida en 61 o 103 hexágonos, de mayor superficie cuanto más periféricos, que alternan de blanco al negro de forma pseudo-aleatorizada.
- El valor de la luminancia en la fase de luz será al menos de 100cd/m2 con un contraste >90%.
Filtros: las normas internacionales aconsejan banda pasante de 5-200Hz. Filtro paso alto 3-10Hz; filtro paso bajo 100-300Hz.
Respuesta del ERG-mf
- La respuesta de primer orden (kernel K1) es una onda bifásica con un componente negativo inicial (N1) seguido de un pico positivo (P1). A veces, puede verse una segunda onda negativa (N2).
- Es una respuesta parecida a la onda del ERG fotópico convencional: la onda N1 está producida por los fotorreceptores y la P1 básicamente por las células bipolares.
Al tratarse de una prueba topográfica, en lugar de amplitudes y latencias de la onda, obtenemos la densidad de la respuesta por unidades de superficie retiniana en cada hexágono, expresada en nanovoltios por grado al cuadrado.
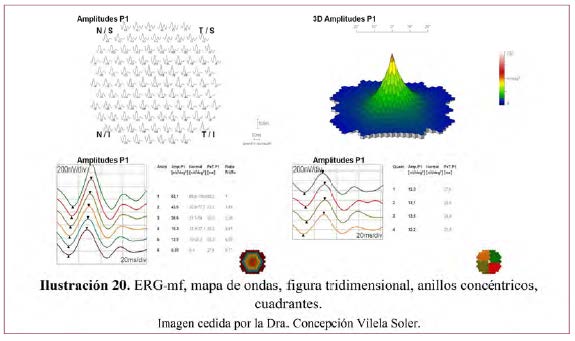
- Los resultados se pueden presentar como un mapa de ondas en el que cada onda representa la actividad de un área del polo posterior o por grupos, que pueden ser (Ilustración 20):
- Anillos concéntricos alrededor de la fóvea
- Cuadrantes
- Una figura tridimensional
Interpretación del ERG–mf
Patrones de afectación:
- Con el ERG-mf se puede valorar la función de todas las capas de la retina si se consideran las ondas de primer y segundo orden.
- En afecciones de los 30-50º centrales de la retina podemos encontrar alteración de la morfología del mapa de ondas, disminución de la densidad de amplitudes por anillos y/o por cuadrantes y alteración de la morfología en 3D.
- En cataratas y en presencia de errores refractivos estará alterado.
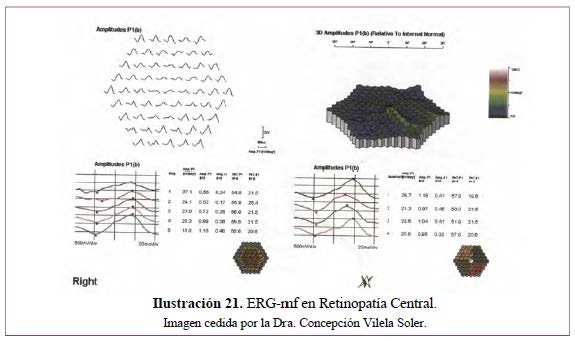
RETINOPATÍA CENTRAL (Ilustración 21)
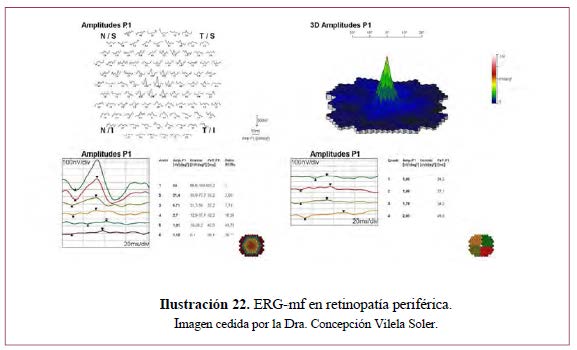
RETINOPATÍA PERIFÉRICA
Aplicaciones clínicas del ERG-mf
- Valorar la función macular, diferenciando maculopatías de la patología del nervio óptico, cuando el FO es normal.
- Ejemplos: casos de pérdida de visión inexplicable sobre todo asociado al estudio de los PEVP y ERG-P, en las que si aparece una alteración del PVE junto con abolición de la N95 del ERG-P con ERG-mf normal pensaremos en enfermedad del nervio óptico
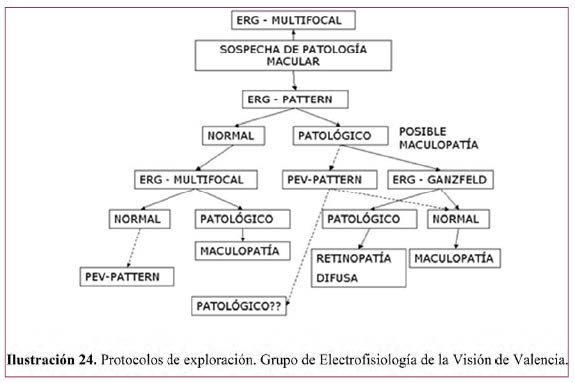
- Diagnóstico y seguimiento de patología macular. También puede permitir evaluar o comparar la respuesta terapéutica en maculopatías (Ilustración 24).
- Ejemplos: Distrofia macular oculta, degeneración macular asociada a la edad, edema macular, traumatismos, cirugía y otros tratamientos retinianos
- Diagnóstico de enfermedades hereditarias extensas de la retina, como complemento del ERG-G. Da información de la función visual y pronóstico de la función macular (Ilustración 21 e Ilustración 22).
- Monitorizar la toxicidad de fármacos sistémicos: cloroquina e hidroxicloroquina.113,114
- Útil en comorbilidad, comparándolo con el CV, permite separar el componente retiniano de otros.
C. Electrooculograma (EOG)96
EOG es un término introducido por Marg en 1951.101 Mide un potencial eléctrico de reposo de aproximadamente 6 mV que existe entre la córnea positiva y la parte posterior del ojo negativa. El ojo normal actúa como una batería, con una diferencia de voltaje entre la córnea y la parte posterior del globo.
El EOG es el registro del potencial de reposo ocular que depende, fundamentalmente, del estado funcional del EPR. Este potencial de reposo varía según las condiciones de iluminación, siempre que no esté alterado el complejo formado por el segmento externo del fotorreceptor y el EPR.
La medida del EOG se realiza de forma indirecta por las variaciones que presenta con el desplazamiento ocular.
La aplicación clínica del EOG es el estudio de las enfermedades del EPR y del fotorreceptor.
Arden y Fojas demuestran que la información más fiable no se refiere a valores absolutos de amplitudes del potencial sino a la comparación de la amplitud con luz/amplitud en oscuridad. En 1962 Arden, Barrada y Kelsey115-117 instauran un test para medir indirectamente el aumento del potencial de reposo como indicador de la función del EPR. El resultado de este test se expresa como Índice de Arden.117
El EPR tiene tres respuestas conocidas:
- La onda c del ERG-G: debido a la hiperpolarización de fotorreceptores frente al estímulo luminoso
- Las ondas rápidas (o pico de oscuridad) de EOG: hiperpolarización tardía de membrana basal del EPR
- Las ondas lentas (o pico de luz) del EOG: despolarización lenta de la membrana basal del EPR
La luz condiciona una polarización de la membrana basal del EPR. Esto se traduce en cambios del potencial transepitelial. El EOG clínico mide la amplitud del potencial de reposo y de la respuesta a la luz.
El EOG tiene dos componentes: uno sin luz y otro con luz. La respuesta sin luz del EOG se relaciona con la integridad del EPR, además de otras fuentes extrarretinianas como la córnea, el cristalino y el cuerpo ciliar. Es independiente del funcionamiento de los fotorreceptores.
El componente sensible a la luz o respuesta lenta, se genera en la membrana basal del EPR, relacionado con cambios en la concentración de potasio. La integridad de los fotorreceptores es necesaria para generarla. Además, esta respuesta requiere el contacto físico entre fotorreceptores y EPR, que se rompería por ejemplo en el DR. También se ha relacionado con esta respuesta la contribución de la capa nuclear interna. Un pico normal de luz requiere fotorreceptores funcionando normalmente y en contacto con el EPR.
La ISCEV propone un protocolo para dos métodos alternativos96: la relación Pico de luz/Pico oscuridad=Índice de Arden. La relación pico de luz con línea de base adaptada a oscuridad. Se puede hacer con dilatación o sin dilatación, las normas de la ISCEV dan a escoger, advirtiendo que se necesita más tiempo dilatado.
Para su registro hay que utilizar electrodos fuera del globo ocular, en los cantos externo e interno del ojo, para favorecer el libre movimiento del mismo.
Se recomienda un estimulador de «campo completo» (Ganzfeld), ya que el EOG es una respuesta en masa de la retina. Se debe utilizar un punto de fijación que induzca un movimiento ocular con un ángulo visual de aproximadamente 30º en el meridiano horizontal.
El paciente debe mover los ojos de derecha a izquierda mientras se monitoriza la actividad eléctrica, observando un cambio brusco de la línea de registro del potencial con cada movimiento. Primero se realiza en condiciones de oscuridad y luego con iluminación, aumentado con el tiempo la diferencia del potencial.
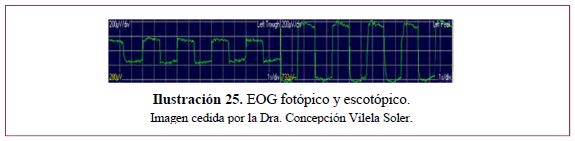
Índice de Arden (Ilustración 25).
Sin dilatación pupilar. Tiene tres fases:
- Pre-adaptación: 15 minutos con luz de habitación
- Fase de oscuridad: 15 minutos en oscuridad. La amplitud mínima aparece a los 11-12 minutos
- Respuesta a la luz: con la luz dada, se recoge EOG hasta que se vea el pico de luz, en 6-9 min
El Índice de Arden se obtiene dividiendo el valor de la amplitud máxima fotópica por la amplitud mínima escotópica y es normal el valor de 2,0 o más. Cada laboratorio debe tener sus propios valores normales. Algunos investigadores consideran valores normales a partir de 1,80; de 1,65 a 1,80 parcialmente normal y alterado por debajo de 1,65.
Aplicaciones clínicas del EOG
Cualquier enfermedad que afecte a la función de los bastones, afectará al EOG. El pico luz está muy afectado en RP y otras degeneraciones de fotorreceptores bastones.
Según G. Arden,116,117 el EOG clínico es útil para:
- El diagnóstico de la distrofia viteliforme macular de Best se considera casi patognomónico de esta enfermedad, encontrar un EOG alterado con un ERG-G normal y encontrar anomalías en portadores.
- El diagnóstico de distrofias de fotorreceptores especialmente de bastones en las que con afectación extensa de retina encontraremos alterados EOG y ERG (RP).
- Conocer la integridad de la función del EPR y sus conexiones con la capa de fotorreceptores.
- Seguimiento de toxicidad de medicamento como antipalúdicos de síntesis, vigabatrina, didanosina, deferoxamina y neurolépticos como la clorpromazina o la tioridazina son fármacos que han demostrado efectos tóxicos sobre la retina en algunos pacientes con tratamientos prolongados. El EOG puede presentar anomalías en estadios preclínicos de intoxicación, siendo un arma valiosa para indicar la interrupción o reducción del tratamiento.
Resumen de la evidencia
| Calidad muy baja | El PEV con diferentes estímulos (damero o haz de luz) es la herramienta para el estudio del nervio óptico, cintilla óptica y resto de la vía visual hasta corteza occipital.94 |
| Calidad muy baja | El ERG-G estudia la retina globalmente y diferencia patología de conos y de bastones. También da información de la función de distintas capas de la retina (externa o interna).93 |
| Calidad muy baja | El ERG-P es la prueba de elección para el estudio de la retina central, mácula y para diferenciar enfermedad macular de enfermedad del nervio óptico.97 |
| Calidad muy baja | El ERG-mf realiza un mapa topográfico de los 30-50º centrales de la retina. Es útil para el estudio de retinopatías sobre todo centrales, pero también para seguimiento de afectación central cuando progresan las periféricas.95 |
| Calidad muy baja | El EOG es el registro del potencial de reposo ocular que depende de la integridad del EPR y los fotorreceptores. Se utilizará en el estudio de Enfermedad de Best fundamentalmente.96 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- No se identificaron estudios que hayan analizado los costes y usos de recursos o sobre valores y preferencias de los pacientes en relación a esta pregunta.
- Aceptabilidad: en el estudio cualitativo con pacientes con DHR en el contexto español, los participantes consideraron estas pruebas como molestas pero aceptables, especialmente si tienen una finalidad diagnóstica. Expresaron determinadas barreras a la hora de someterse a las pruebas que se traducen en las siguientes demandas:
- Que se facilite el acceso a las pruebas (por ejemplo, evitar el uso de pantallas con número para pasar a un servicio, ya que éstas no están adaptadas a los pacientes con baja visión o sin visión puesto que no son parlantes).
- Que se adapte el lenguaje usado (por ejemplo, recibir referencias espaciales verbalmente específicas -a su derecha, delante, detrás, etc.-, en lugar de gestuales).
- Que el profesional sanitario acompañe a los pacientes en los traslados entre consultas y acceso a las pruebas, especialmente si la persona no va acompañada por un familiar o conocido.
- Que se realicen pruebas adaptadas a las condiciones específicas de baja visión de cada patología y a su resto visual.
- Factibilidad: la electrofisiología no está disponible en todos los hospitales de referencia.
El grupo elaborador formuló las siguientes recomendaciones de buena práctica considerando las opciones que podrían ayudar al diagnóstico y confirmación de las DHR.
Recomendaciones
| √ | Se recomienda seguir los estándares de la Sociedad Internacional de Electrofisiología Clínica y Visión (ISCEV) para la realización de todas y cada una de las pruebas electrofisiológicas. |
| √ | Se sugiere la supervisión por parte de un médico formado en electrofisiología de la visión, bien un oftalmólogo o bien un neurofisiólogo, del estudio electrofisiológico que será quien emita el informe correspondiente. |
| √ | Si la enfermedad afecta al nervio óptico y/o la vía visual, se sugiere solicitar un potencial evocado visual. |
| √ | Si la patología afecta al EPR, se sugiere solicitar un electrooculograma. |
| √ | Si se desea estudiar la retina, se sugiere solicitar un electrorretinograma (ERG), teniendo en cuenta que existe la posibilidad de estudiar retina central con el ERGpattern, diferenciar conos y bastones con el ERG-Ganzfeld o de campo lleno, y realizar un mapa de los 30 grados centrales de la retina con el ERG multifocal. |
5.2.3.1. Retinosis pigmentaria (RP)
La etapa de aparición es, frecuentemente, la adolescencia. Los hallazgos clínicos más frecuentemente observados en la RP son: ceguera nocturna o nictalopía, fotofobia, reducción concéntrica del CV o visión en túnel, migración intrarretiniana de pigmento desde la retina externa, ERG de bastones disminuido o abolido (disminución importante en las amplitudes de las ondas a y b) con ERG de conos inicialmente conservado pero que en etapas tardías también puede estar afectado, además de atrofia progresiva de la retina externa, palidez del nervio óptico por gliosis, atenuación de los vasos retinianos (quizás el signo de aparición más precoz) y, eventualmente, pérdida de visión central en las etapas más tardías.21
muy baja
Síndrome de Usher
Se han descrito tres subtipos clínicos denominados USH1, USH2 y USH3 de acuerdo con la gravedad del deterioro auditivo, la presencia o ausencia de disfunción vestibular, y la edad de inicio de la RP:22
- USH1: déficit auditivo congénito profundo con disfunción vestibular severa y diagnóstico pre-puberal de la RP.
- USH2: déficit auditivo congénito moderado a profundo no progresivo, con ausencia de disfunción vestibular y diagnóstico postpuberal de la RP.
- USH3: déficit auditivo moderado y progresivo que aparece, generalmente en la 1ª década, con ausencia o presencia de disfunción vestibular. El diagnóstico de RP puede ser pre- o post-puberal.
La disfunción visual en los síndromes de USH1 y USH2 se diagnostica en promedio en las edades de 17 y 24 años, respectivamente. El diagnóstico tardío tiene consecuencias perjudiciales para las personas sordas con USH1. De hecho, estos pacientes, pueden beneficiar desde el principio de los implantes cocleares (prótesis con arrays de electrodos que estimulan directamente las neuronas auditivas primarias), para la adquisición del lenguaje oral, si el diagnóstico es precoz. Los datos clínicos precisos, apoyados por el diagnóstico molecular, son fundamentales para el asesoramiento genético, orientación educativa y el inicio de estrategias terapéuticas para el USH.22 En este sentido, mientras que las aproximaciones de genotipado utilizando microarrays han detectado mutaciones sólo en un 30-50% de estos pacientes, las aproximaciones en las que se utiliza la secuenciación de todo el exoma mejora claramente la eficacia del diagnóstico molecular para las mutaciones que se identifican en 90% de los casos.118
muy baja
Síndrome de Bardet-Biedl
El fenotipo es característico y asocia a RP o más a menudo a una CORD, con obesidad presente desde la infancia, discapacidad intelectual o retraso psicomotor leve, polidactilia postaxial, hipogonadismo y anomalías renales que conducen a la insuficiencia renal.23,24
muy baja
5.2.3.2. Distrofia progresiva de conos (PCD) y distrofia de conos y bastones (CORD)
Las PCD normalmente no dan síntomas hasta la adolescencia o el inicio de la edad adulta. La edad de comienzo de la pérdida de visión, así como la tasa de progresión muestran una gran variabilidad entre las distintas familias, aunque habitualmente la visión baja hasta niveles de 20/200. La fotofobia es a menudo el síntoma más precoz y prominente. Además, normalmente la visión de colores está afectada desde el inicio de la enfermedad y a menudo progresa a una ceguera completa de colores. En el FO suele hallarse la típica maculopatía en ojo de buey con periferia normal que es mucho más evidente en la AFG o la autofluorescencia. Los nervios ópticos pueden mostrar o no atrofia temporal.45 En el CV suelen mostrar escotoma central. Existe un deterioro progresivo en la respuesta de amplitud de los conos, mientras que los registros de los bastones, inicialmente, permanecen normales en el ERG.24
Por su parte, los pacientes que presentan fenotipo de CORD desarrollan los hallazgos típicos de una PCD recién descritos al principio de su vida, pero no tardando mucho aparece la afectación de bastones y por lo tanto la ceguera nocturna. La afectación retiniana normalmente es el único hallazgo, pero, en algunos casos, se asocia a otras anomalías sistémicas constituyéndose en formas sindrómicas (Para más información, ver 5.2.4. Diagnóstico de las distrofias sindrómicas). En los pacientes con CORD, el FO muestra atrofia macular o una maculopatía en ojo de buey en estadíos precoces. Más tarde puede aparecer atrofia del EPR periférico, pigmentación retiniana en espículas, atenuación arterial y palidez del nervio óptico.21 El CV muestra un escotoma central y compromiso variado del CV periférico. El ERG muestra repuestas reducidas para conos y bastones, más severamente comprometida para los conos.24
La edad media de ceguera legal es de 48 años en los casos de PCD y de 35 años en los casos de CORD. Sin embargo, los pacientes con mutaciones en el gen ABCA4 (9% de los pacientes con PCD AR y 26% de individuos con CORD AR) y con edad de inicio inferior a los 20 años, presentan una progresión más rápida hacia la ceguera legal. Así, ambos trastornos evolucionan progresivamente hacia la ceguera legal en la mayoría de los pacientes.46
muy baja
5.2.3.3. Enfermedad de Stargardt y fundus flavimaculatus
La edad de inicio es típicamente entre 10 y 20 años. Sin embargo, las alteraciones pueden aparecer en una edad más precoz e incluso a edad más tardía (> 45 años)119 y asociarse con un mejor pronóstico visual.120 Clínicamente puede tener varias formas de presentación aunque a veces un mismo paciente pasa de una de estas formas a otras a lo largo de su vida. Estas se dividen en:121
- Fondo ocular color bronce y silencio coroideo: se caracteriza por la presencia de cambios variables en la pigmentación del área macular, aspecto de «metal batido» con atrofia eventual del EPR y de la coriocapilar. Posteriormente aparece un anillo de flecks que a menudo respeta la fóvea en un área dentro de un diámetro de disco.122 Inicialmente, los pacientes refieren escotomas relativos centrales o paracentrales. En la AFG se observa el silencio coroideo casi patognomónico de esta enfermedad (sólo visible también en casos de intoxicación por plata). El ERG-G y el EOG no suelen alterarse, pero sí se pueden ver alteraciones en el ERG-mf.
- Fundus Flavimaculatus: se caracteriza por la presencia de flecks que salen del polo posterior. Las manchas pueden extenderse más allá de las arcadas vasculares y, a menudo, del lado nasal del disco óptico. En esta etapa puede haber una reabsorción parcial de las manchas de mayor tiempo de evolución. Los CV periféricos son normales, y puede producirse un escotoma central relativo. Si la retina está muy alterada por la presencia de numerosos flecks se puede buscar el silencio coroideo de la AFG en la retina peripapilar que típicamente está bastante bien preservada. Las amplitudes del ERG y las ratios del EOG pueden presentar registros normales o subnormales pero se pueden ver alteraciones en el ERG-P o en el ERG-mf. Estos pacientes manifiestan períodos prolongados para la adaptación a la oscuridad.
- Maculopatía atrófica con o sin flecks amarillentos: caracterizado por la presencia de flecks difusamente (algunos de ellos reabsorbidos) asociados a la aparición de áreas de atrofia del EPR y la coriocapilar en el área macular. El ERG y EOG pueden estar reducidos pero son más marcadas las alteraciones en el ERG-P o en el ERG-mf, con tiempos prolongados de adaptación a la oscuridad. Los defectos de CV central son similares a los observados en el estadio 2 con deterioro evidente de los campos periféricos.
- Maculopatía atrófica con signos y síntomas tardíos de afectación de bastones: caracterizado por la presencia de flecks difusamente reabsorbidos y extensas áreas de atrofia del EPR y la coriocapilar en todo fondo ocular. Hay constricción moderada o acentuada de los CV periféricos. El ERG se presenta notablemente reducidos en las amplitudes de la respuesta tanto para conos como para bastones. Estos pacientes presentan tiempos muy prolongados de adaptación a la oscuridad.
muy baja
5.2.3.4. Ceguera nocturna congénita estacionaria
La ceguera nocturna congénita no progresiva o estacionaria se caracteriza por el comienzo de nictalopía en la infancia que no progresa y con buena AV. Algunas formas pueden asociarse a baja AV, nistagmus y estrabismo. Muestran ERG negativo o decapitado en el ERG-G mixto en condiciones escotópicas. La adaptación a la oscuridad puede estar ausente o se encuentra muy prolongada y suelen ser miopes.38-44
Se pueden subdividir en dos grupos, los que tienen FO normal que son mucho más frecuentes y los que tienen anormalidades claras de fondo. Las que cursan con fondo anormal son el Fundus Albipunctatus (manchas blanquecinas en periferia media) y la Enfermedad de Oguchi (fondo dorado que adquiere coloración normal en oscuridad).
muy baja
5.2.3.5. Distrofia viteliforme macular de Best o enfermedad de Best
La distrofia viteliforme macular de Best es una forma lenta y progresiva de distrofia macular. En la mayoría de los casos tiene su comienzo en la infancia, aunque a veces se puede desarrollar en edad más avanzada. Aunque esta enfermedad se considere la segunda forma más común de maculopatía juvenil, con un inicio usualmente antes de los 15 años de edad, sólo alrededor del 1% de todos los casos de maculopatía pueden atribuirse a la enfermedad de Best.60
muy baja
La distrofia viteliforme macular de Best se caracteriza por síntomas de metamorfopsia, visión borrosa, y la disminución de la visión central. En el FO se observa una lesión macular en «yema de huevo» bien circunscrita en los estadíos más precoces.123,124
muy baja
La evolución oftalmoscópica de la lesión macular obedece a cuatro etapas,70 la OCT y la autofluorescencia contribuye para la clasificación de la lesión macular:125
La etapa primaria se describe por el FO normal (pre-viteliforme) con tan solo un EOG alterado y ERG normal lo que se considera patognomónico de esta enfermedad.
La etapa de «yema de huevo» la lesión macular es típica con color amarillento, redondeada y bien delimitada, algo elevada y ubicada, generalmente, en el centro del área macular (viteliforme y pseudo-hipopion).
En la etapa siguiente, la lesión macular asume un aspecto de «huevos revueltos» debido a la rotura y desestructuración de la lesión (vitelorruptiva).
La etapa final se caracteriza por una cicatriz hipertrófica (cicatricial), maculopatía atrófica, o bien una cicatriz fibrovascular que generalmente es secundaria a una neovascularización coroidea frecuente en esta enfermedad.70
Sin embargo, el grado de deterioro de la visión central y la edad de aparición de los síntomas varían ampliamente, incluso entre miembros de la misma familia. Los pacientes tienen la visión periférica y adaptación a la oscuridad normales. La combinación de un EOG anormal con ERG-G normal es una característica peculiar de esta enfermedad.60 No obstante, el ERG-mf puede mostrar amplitudes reducidas en las áreas centrales, incluso durante las primeras etapas de la afectación macular y el ERG-P puede estar también alterado.
muy baja
5.2.3.6. Retinosquisis juvenil ligada a X (XLRS)
Clásicamente se caracteriza por la pérdida de visión de inicio temprano debido a esquisis foveal en ambos ojos. El patrón macular clásico en «radios de rueda de bicicleta» que se irradian hacia fuera del área foveal. Aproximadamente la mitad de los pacientes asocian también retinosquisis periférica que suele ser inferior. Las retinosquisis periféricas ocasionan un aumento del riesgo de DR que se estima afecta a un 5-20% de los pacientes, también una tercera parte de estos enfermos pueden sufrir hemorragias en la cavidad quística o en la cavidad vítrea. Algunos pacientes pueden mostrar estrechamiento progresivo de los vasos retinianos y aparición de cambios pigmentarios periféricos dando un aspecto similar a la RP. Hallazgos adicionales incluyen manchas blancas difusas de la retina que se asemejan a las del fundus albipunctatus, envainamiento perivascular, vasos dendritiformes en la retina periférica, velos vítreos, algunos de los cuales resultan de la separación de la fina capa interna de la cavidad de la esquisis, y más raramente retinopatía exudativa como en la enfermedad de Coats. El trastorno se puede presentar en la infancia temprana con estrabismo, nistagmus, hipermetropía axial, ectopia foveal, o hemorragia en la cavidad de la esquisis o en el vítreo.66
muy baja
La OCT ayuda a visualizar las características patológicas maculares en la XLRS. El ERG clásicamente demuestra la reducción selectiva de la onda b, mientras que la onda a permanece dentro de un rango normal lo que da lugar a la típica aparición de un ERG negativo importante para el diagnóstico.124
A veces, en la edad adulta se observa la desaparición de los cambios quísticos del área macular, con aparición de una alteración más extensa del EPR subyacente, y finalmente desarrollo de una lesión atrófica macular inespecífica.126,127
muy baja
5.2.3.7. Coroideremia
Los síntomas típicos de la coroideremia incluyen ceguera nocturna y constricción concéntrica del CV desde la 1ª ó 2ª década de la vida, por lo que puede ser confundida con RP. La visión central generalmente se conserva hasta la década de los 50, lo que puede ayudarnos a distinguirla de la XLRP en que se suele afectar mucho más pronto en la vida. Las características fundoscópicas típicas cuando la enfermedad progresa incluyen pérdida total de pigmento tanto a nivel del EPR como de la coriocapilar, acompañada de una pérdida extensa de la capa coriocapilar, en este momento el aspecto de fondo es típico pues se visualizan claramente los grandes vasos coroideos y ya no se confunde tan fácilmente con la RP.23,128
muy baja
Aunque las mujeres portadoras suelen tener un fenotipo muy típico, que puede ayudar a su detección cuando el diagnóstico genético molecular no está disponible, un FO normal en una mujer con antecedentes de varones con coroideremia, no excluye el diagnóstico de «portadora». Las portadoras pueden tener sólo cambios pigmentarios leves en media periferia, o presentar degeneración del EPR y la coroides en parches más extensos de la retina, o incluso presentar un ojo afecto y el otro prácticamente normal. Por otra parte, puede haber cambios peripapilares degenerativos y depósitos drusenoides blanquecinos en la media periferia, o alternativamente la apariencia puede ser muy similar a la de los varones afectos. Los cambios del EPR de las portadoras son más evidentes en la autofluorescencia. La electrofisiología en pacientes afectos es similar a la de la RP. Sin embargo, la electrofisiología en las portadoras de coroideremia suele ser completamente normal, al contrario de lo que sucede con las portadoras del XLRP en las que suele encontrarse alguna anomalía, ayudando también a veces a diferenciar ambas enfermedades.28,29,31
muy baja
La atrofia girata suele comenzar con nictalopía en la primera década de la vida seguida de pérdida de CV periférico y también pérdida de AV, por lo tanto, la sintomatología es indistinguible de la RP. Es frecuente encontrar miopía elevada, astigmatismo, alteraciones en el patrón de autofluorescencia y catarata subcapsular posterior. En el FO se observan parches circulares de atrofia coroidea confluentes en la periferia que se extienden desde allí hacia la retina más anterior. Algunos de los grandes vasos coroideos pueden estar conservados. Las lesiones permanecen separadas entre sí por bordes finos de pigmento que le dan un aspecto festoneado muy característico a la periferia de la retina. La mácula se hace anormal en estadíos muy tardíos tanto en varones afectos como en mujeres portadoras. Las determinaciones analíticas (encontrar una elevación de ornitina en el plasma o la orina confirma el diagnóstico)36 permite de modo inequívoco el diagnóstico diferencial entre atrofia girata y coroideremia o RP.32-36
muy baja
Es la forma más precoz y la más severa de todas las DHR, responsables de los casos de ceguera congénita Los criterios diagnósticos actualmente reconocido, aunque todavía debatidos, para la identificación de la ACL son:129 a) inicio temprano de ceguera o visión deficiente, que aparece en el primer año de vida, antes de los seis meses de edad; b) reflejos pupilares lentos; c) nistagmus o movimientos oculares erráticos; d) signo óculo-digital (los niños se frotan o se presionan los ojos con frecuencia buscando ocasionarse fotopsias); e) ERG fotópico y escotópico abolidos o severamente reducido; f) PEV ausentes o anormales; g) aspecto del FO variable (normal en las fases más incipientes, marmóreo, y después albinótico, con pigmentación, etc.). Estos pacientes suelen ser hipermétropes a diferencia de otras DHR.
Aunque es muy raro, además de estos síntomas oculares se han descrito asociaciones de forma variable, que incluye: retardo del desarrollo neuropsíquico y motor, retraso mental, y otras anomalías sistémicas asociadas. El diagnóstico diferencial de esta enfermedad se complica por el hecho de que varios autores han descrito hallazgos oculares típicos de ACL asociados a síndromes genéticos y en síndromes metabólicos o enfermedades degenerativas del sistema nervioso.26
muy baja
Resumen de la evidencia
| Retinosis Pigmentaria | |
| Calidad muy baja | El cuadro de la RP se caracteriza por ceguera nocturna o nictalopía de aparición precoz, visión en túnel, migración intrarretiniana de pigmento desde la retina externa, ERG de bastones disminuido o abolido, atrofia progresiva de la retina externa, velos vítreos, palidez del nervio óptico y atenuación de los vasos retinianos.21 |
| Síndrome de Usher | |
| Calidad muy baja | El cuadro de USH se caracteriza por trastornos combinados de sordera-ceguera con tres subtipos clínicos denominados USH1, USH2 y USH3 de acuerdo con la gravedad del deterioro auditivo, la presencia o ausencia de disfunción vestibular, y la edad de inicio de la RP.22
|
| Síndrome de Bardet-Biedl | |
| Calidad muy baja | El síndrome de Bardet-Biedl asocia RP, o más frecuentemente distrofia de conos y bastones, con obesidad presente en la infancia, discapacidad intelectualo retraso psicomotor leve, polidactilia axial, hipogenitalismo y anomalías renales que conducen a la insuficiencia renal.23,24 |
| Distrofia Progresiva de Conos y Distrofia de Conos y Bastones | |
| Calidad muy baja | Las PCD normalmente no dan síntomas hasta la adolescencia. La visión baja hasta niveles de 20/200. La fotofobia es el síntoma más precoz y prominente. Suelen presentar visión de colores afectada que progresa a una ceguera completa de colores, maculopatía en ojo de buey con periferia normal y los nervios ópticos pueden mostrar o no atrofia temporal.45 |
| Calidad muy baja | Las PCD en el CV muestran escotoma central. En el ERG presentan deterioro progresivo en la respuesta de amplitud de los conos, mientras que, inicialmente, los registros de los bastones permanecen normales.24 |
| Calidad muy baja | Los pacientes con CORD desarrollan los hallazgos de una PCD al principio de su vida, el FO muestra atrofia macular en ojo de buey en estadíos precoces y más tarde puede aparecer atrofia del EPR periférico, pigmentación retiniana en espículas, atenuación arterial y palidez del nervio óptico desarrollando síntomas y signos similares a los de la RP.3 |
| Calidad muy baja | En las CORD el CV muestra un escotoma central y compromiso variado del CV periférico. El ERG muestra repuestas reducidas para conos y bastones, más comprometida para los conos.24 |
| Enfermedad de Stargardt y fundus flavimaculatus | |
| Calidad muy baja | La edad de inicio es muy variable desde la primera a la quinta década de la vida, aunque típicamente la enfermedad comienza entre los 10 y los 20 años.120 |
| Calidad muy baja | Clínicamente puede dividirse en:121
|
| Ceguera Nocturna Congénita Estacionaria | |
| Calidad muy baja | La ceguera nocturna congénita no progresiva o estacionaria se caracteriza por el comienzo de nictalopia en la infancia que no progresa y con buena AV, puede asociarse a baja AV, nistagmo y estrabismo. Es importante la detección de un ERG-G escotópico mixto negativo en el diagnóstico. La adaptación a la oscuridad puede estar ausente o se encuentra muy prolongada y suelen ser miopes.38-44 |
| Se pueden subdividir en dos grupos, los que tienen FO normal (los más frecuentes) y los que tienen anormalidades claras de fondo (Fundus Albipunctatus y la Enfermedad de Oguchi). | |
| Distrofia Viteliforme Macular de Best o enfermedad de Best | |
| La distrofia viteliforme macular de Best es una forma lenta y progresiva de distrofia macular con comienzo normalmente en la infancia, salvo que se complique como hace frecuentemente con una membrana neovascular subretiniana, en cuyo caso produce una pérdida más o menos brusca de visión con agudización de la metamorfopsia. | |
| Calidad muy baja | Los pacientes suelen presentar visión periférica y adaptación a la oscuridad normales. Lo que resulta casi patognomónico para el diagnóstico es encontrar un EOG anormal con ERG clásico normal.60 No obstante, el ERG-mf puede mostrar amplitudes reducidas en las áreas centrales. |
| Calidad muy baja | La distrofia viteliforme macular de Best se caracteriza por síntomas de metamorfopsia, visión borrosa, y la disminución de la visión central. En el FO es muy característica la presencia de una lesión macular en «yema de huevo» bien circunscrita en la primera década de la vida.123,124 |
La evolución oftalmoscópica de la lesión macular obedece a cuatro etapas,70 la OCT contribuye para la clasificación de la lesión macular:125
Sin embargo, es importante recordar que hay cuadros de Best multifocal en los que no hay una sola lesión, sino varias lesiones en yema de huevo dispersas en el FO. |
|
| Retinosquisis Juvenil ligada a X | |
| Calidad muy baja | La XLRS se caracteriza por la pérdida de visión de inicio temprano y esquisis foveal en ambos ojos, patrón macular en «radios de rueda» que se irradian hacia fuera del área foveal. La esquisis puede afectar sólo al área foveal, o también a la retina periférica (50% de los pacientes). En ocasiones, se pueden ver adicionalmente manchas blancas difusas de la retina, vasos dendritiformes en la retina periférica, y velos vítreos.66 |
| Calidad muy baja | Si existe esquisis periférica suele ser inferior y da lugar a un aumento del riesgo de DR (5-20%), también pueden sufrir hemorragias en la cavidad quística o en la cavidad vítrea (1/3).66 |
| Calidad muy baja | El trastorno se puede presentar en la infancia con estrabismo, nistagmus, hipermetropía axial, ectopia foveal, o hemorragia en la cavidad de la esquisis o en el vítreo.66 |
| Calidad muy baja | El ERG muestra la reducción selectiva de la onda b, mientras que la onda a permanece normal, lo que da lugar a la típica aparición de un ERG negativo siempre que la afectación sea lo suficientemente extensa.124 |
| Calidad baja |
A veces, en la edad adulta pueden desaparecer de los cambios quísticos, con aparición de una alteración del EPR subyacente y desarrollo de una lesión atrófica macular inespecífica.126,127 |
| Coroideremia | |
| Calidad muy baja | Los síntomas de la coroideremia incluyen ceguera nocturna y constricción concéntrica del CV desde la 1ª ó 2ª década de la vida, visión central conservada hasta la década de los 50. Las características fundoscópicas típicas incluyen pérdida total de pigmento, acompañada de una pérdida extensa de la capa coriocapilar.23,128,130 |
| Calidad muy baja | Las portadoras pueden tener sólo cambios pigmentarios leves en media periferia, o presentar degeneración del EPR y la coroides en parches más extensos de la retina; cambios peripapilares degenerativos y depósitos drusenoides blanquecinos en la media periferia.28-31 |
| Calidad muy baja | La electrofisiología en pacientes es similar a la de la RP. Sin embargo, la electrofisiología en las portadoras suele ser completamente normal, al contrario que en las portadoras de XLRP, lo que ayuda a diferenciar ambas enfermedades. |
| Atrofia girata | |
| Calidad muy baja | La atrofia girata comienza con nictalopía en la primera década de la vida seguida de pérdida de CV periférico y de AV, con miopía elevada, astigmatismo y catarata subcapsular. En el FO se observan parches circulares de atrofia coroidea confluentes en la periferia que se extienden desde la retina anterior a la posterior. Las lesiones permanecen separadas entre sí por bordes finos de pigmento que le dan un aspecto festoneado a la periferia de la retina muy característico de la enfermedad.32-36 |
| Amaurosis congénita de Leber (ACL) | |
| Calidad muy baja | La ACL es la forma más temprana y la más severa de todas las DHR, responsables de los casos de ceguera congénita. El cuadro de la ACL se caracteriza por inicio temprano de la ceguera, reflejos pupilares lentos, nistagmus, signos óculo-digitales como frotarse los ojos, ERG fotópico y escotópico abolidos o severamente reducido y PEV ausentes o anormales.129 A diferencia de otras DHR, los pacientes suelen ser hipermétropes. |
| Calidad muy baja | El aspecto del FO es variable (normal al inicio, marmóreo, albinótico con pigmentación, etc.).129 |
| Calidad muy baja | Se han descrito asociaciones de ACL con retardo del desarrollo neuro-psíquico y motor, retraso mental, y otras anomalías sistémicas asociadas.26 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
Calidad global de la evidencia: la evidencia consiste en aproximaciones descriptivas, cohortes reducidas, series de casos y casos aislados por lo que la calidad global de la evidencia se considera muy baja. En la actualidad, la gran parte de los estudios se centran en la investigación genética y sobre las bases moleculares de la mutación genética relacionada con el objetivo de una mejor comprensión de cuadro clínico.
El grupo elaborador formuló las siguientes recomendaciones considerando las opciones que podrían ayudar al diagnóstico y confirmación de las DHR.
Recomendaciones
| √ | Se sugiere considerar los criterios clínicos básicos con relación al inicio de la aparición de la sintomatología, la caracterización del cuadro clínico actual y de presentación, la historia familiar (la presencia de antecedentes familiares apoya el diagnóstico pero su ausencia no lo descarta) y general del paciente. La orientación diagnóstica se hará según los criterios oftalmológicos (examen oftalmológico general y del FO) y con base a la electrofisiología ocular. La OCT y la autofluorescencia pueden contribuir para la orientación diagnóstica, y a veces también la AFG. La presencia de patología bilateral más o menos simétrica, también debe hacer sospechar patología hereditaria. |
| Retinosis pigmentaria, síndrome de Usher, síndrome de Bardet-Biedl | |
| √ | Para el diagnóstico de la RP, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| √ | Se sugiere considerar la asociación de la RP con hipoacusia neurosensorial asociada o no a trastornos del equilibrio como signos de sospecha para el diagnóstico de USH. |
| √ | Se sugiere considerar la asociación de la RP con polidactilia, alteraciones genitourinarias, del desarrollo neuro-psicomotor y obesidad como signos de sospecha para el diagnóstico de síndrome de Bardet-Biedl. |
| Distrofia progresiva de conos (PCD) y distrofia de conos y bastones (CORD) | |
| √ | Para el diagnóstico de PCD y CORD, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Enfermedad de Stargardt y fundus flavimaculatus | |
| √ | Para el diagnóstico de fundus flavimaculatus y enfermedad de Stargardt, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Ceguera nocturna congénita estacionaria | |
| √ | Para el diagnóstico de la ceguera nocturna congénita no progresiva o estacionaria, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Distrofia viteliforme macular de Best o enfermedad de Best | |
| √ | Para el diagnóstico de la distrofia viteliforme macular de Best, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Retinosquisis juvenil ligada a X (XLRS) | |
| √ | Para el diagnóstico de retinosquisis juvenil ligada a X se sugiere considerar los siguientes signos de sospecha:
|
| Coroideremia | |
| √ | Para el diagnóstico de la coroideremia, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Atrofia girata | |
| √ | Para el diagnóstico de la atrofia girata, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
| Amaurosis congénita de Leber | |
| √ | Para el diagnóstico de la ACL, se sugiere considerar los siguientes signos o síntomas de sospecha:
|
5.2.4. Diagnóstico de las distrofias sindrómicas
- ¿Qué signos y síntomas deben hacernos sospechar que estamos ante una distrofia hereditaria de la retina sindrómica?
Las DHR sindrómicas son cuadros multiorgánicos de tipo congénito hereditario que incluyen, entre otras, alteraciones retinianas. El diagnóstico precoz de estas entidades va a permitir la detección de patologías asociadas potencialmente graves, y su tratamiento precoz podrá prevenir en ocasiones un deterioro funcional irreversible.
Para el desarrollo de esta pregunta se han considerado estudios de series de casos y opiniones de expertos en relación a las DHR sindrómicas más frecuentes.
Existen múltiples síndromes multiorgánicos en que aparecen signos y síntomas oculares similares a los que se observan en los casos aislados de DHR.90,91
Para poder detectar precozmente una DHR sindrómica, debemos estar atentos a la presencia y/o aparición a lo largo de la evolución de signos y síntomas extraoculares. En ocasiones, los hallazgos oftalmológicos pueden ser sutiles y/o aparecer a posteriori.92
Por otra parte, no todos los signos y síntomas sistémicos aparecen en todos los pacientes.131
Síndrome de Usher
baja/muy
baja
Asocia retinopatía pigmentaria progresiva, sordera neurosensorial, alteraciones vestibulares y del equilibrio.
Se clasifica en tres subtipos:
- USH1: Su rasgo diferencial es la ausencia de función vestibular con el consecuente retraso y alteración de la marcha. También asocia sordera profunda neurosensorial preverbal y RP progresiva con afectación visual severa y precoz tanto del CV como de la AV.132 Típicamente presenta hipermetropía sin astigmatismo. A diferencia de la RP no sindrómica, que suele presentar miopía.133
- USH2: Presenta función vestibular normal con disminución de la audición congénita moderada-severa y RP progresiva de aparición tardía y de menor grado que el USH1.134 Puede presentar alteraciones neurorradiológicas.135 Asocian miopía y se asemejan clínicamente a la RP no sindrómica.133
- USH3: Se caracteriza por la disminución progresiva de la audición y de la función vestibular en el periodo postlingual y puede asociar ceguera nocturna de aparición en la edad adulta. Asocian hipermetropía (>2D) con astigmatismo (>1D) clínicamente significativos.133
- Ante todo niños con sordera neurosensorial profunda y retraso de la marcha independiente (debido a una función vestibular alterada), se debe sospechar USH.92
- Múltiples síndromes asocian sordera y degeneración retiniana, aunque el USH es de los más frecuentes. Tal y como concluye el estudio de Mets del año 2000, todos los neonatos con sordera preverbal neurosensorial severa y profunda, deberían ser estudiados para descartar el USH, a través de un examen oftalmológico que incluya test neurofisiológico.134
- Otros autores, también recomiendan los test electroneurofisiológicos auditivos, vestibulares y oftalmológicos para confirmar precozmente el diagnóstico ante la sospecha de USH.92,136,137
- El diagnóstico genético también puede ayudar a confirmar la enfermedad.
baja/muy
baja
En un estudio transversal mediante cuestionario administrado a personas con síndrome de Bardet-Biedl (n=109) y sus familias, se revisaron los criterios para mejorar la detección de este cuadro. Se concluyó que se debe sospechar síndrome de Bardet-Biedl ante la presencia de RP progresiva que no aparece hasta la adolescencia, y que asocia obesidad troncular, hipogonadismo, polidactilia y/o braquisindactilia, ceguera nocturna, retraso psicomotor, del lenguaje y alteraciones del comportamiento.138
Puede asociar también alteraciones renales, diabetes mellitus,139 patología cardiaca, asma, alteraciones faciales, dentales y patologías oftalmológicas como estrabismo, astigmatismo, nistagmo, catarata, discromatopsias y atrofia óptica, entre otras.136,140
Iannaccone, en su estudio de 1997 observó que a diferencia de la RP no sindrómica, la retinopatía en el síndrome de Bardet-Biedl afecta más al área macular por lo que se comporta más bien como una CORD, el nervio óptico presenta más frecuentemente palidez y atrofia, con alteración de los PEV, y se dan más casos de retinopatía pigmentaria sectorial, y de retinopatía sine pigmento. Existe menor correlación entre el tiempo de evolución de la enfermedad y el acúmulo de pigmento, y menor correlación entre la amplitud de la onda b del ERG y las áreas de CV conservadas.131
Por tanto, la ausencia de signos oftalmológicos evidentes en la exploración de un caso con sospecha de DHR, no excluye el diagnóstico. Se recomiendan test de Electroneurofisiología para aproximarnos al diagnóstico.131,137
baja/muy
baja
baja
Presenta coloboma, ceguera, atresia de coanas, malfunción vestibular y auditiva, retraso del crecimiento y del desarrollo psicomotor, déficit intelectual y alteración de pares craneales.141
baja
RETINOPATÍAS EN DISTROFIAS MUSCULARES
Síndrome Kearns Sayre
Es una enfermedad mitocondrial que comienza a dar sus manifestaciones pronto en la vida y que asocia degeneración retiniana (se ha descrito RP típica y también atrofia similar a la que produce la distrofia coroidea areolar central), oftalmoplejia externa progresiva que produce una ptosis palpebral muy severa, fatiga muscular, insuficiencia mitral y alteraciones en el electrocardiograma (bloqueo aurículo-ventricular) entre otras.137,142 Estos pacientes son frecuentemente diabéticos.143
muy baja
Es una enfermedad heredofamiliar caracterizada por miotonía, con atrofia muscular selectiva, calvicie, atrofia testicular y ovárica, senilidad prematura y cataratas. Un 20-25% de estos pacientes muestra evidencia de degeneración retiniana consistente en una variedad de imágenes fundoscópicas como distrofia en patrón con sus distintos patrones y drusas.144 La pérdida de visión suele ser mínima. Las complicaciones oculares en la distrofia miotónica incluyen ptosis, debilidad de los músculos oculares y cataratas. Las cataratas son útiles para el diagnóstico de distrofia miotónica por su elevada frecuencia y aspecto típico. Otras características de la distrofia miotónica incluyen respuesta pupilar lenta, presión intraocular baja, y la degeneración de la retina consistente en depósitos amarillentos trirradiados o pigmentados de configuración pavimentosa en la periferia de la retina. También se ha descrito estrechamiento arteriolar. A veces el ERG muestra respuestas subnormales.145
muy baja
Distrofia muscular de Duchenne
La distrofia muscular de Duchenne es una enfermedad XL que se caracteriza por debilidad muscular proximal progresiva. El 95% de los pacientes habrá perdido la capacidad para caminar a la edad de 12 años y la mortalidad es del 95% a los 20 años. La distrofina, el producto del gen mutado en esta enfermedad se ha encontrado depositada en la capa plexiforme externa de la retina y puede ser importante en la neurotransmisión retiniana. La mayor parte de los pacientes tienen ERG con amplitudes subnormales, hiperpigmentación focal de la mácula; pero son normales la AV, la visión de color y la función de los músculos extraoculares.146
muy baja
Síndrome Cerebro-Hepato-Renal o Síndrome de Zellweger
El síndrome de Zellweger es una miopatía mitocondrial y la variante más grave de los trastornos de la biogénesis del peroxisoma que constituyen el espectro del síndrome Zellweger de herencia AR y que causa acúmulo de los ácidos grasos de cadena larga.147 Este espectro es un continuo de tres fenotipos: 1) el síndrome de Zellweger, la más grave; 2) la adrenoleucodistrofia neonatal; y 3) la enfermedad de Refsum infantil, la menos grave. Todos ellos fueron originalmente descritos antes que las bases bioquímicas y moleculares de estos trastornos se hubieran determinado completamente.
Varios trastornos multisistémicos en la infancia con prominentes manifestaciones oftalmológicas se han atribuido al mal funcionamiento del peroxisoma, un orgánulo subcelular. Los trastornos peroxisomales se han dividido en tres grupos:
- Los que resultan de la biogénesis defectuosa de la peroxisoma: el síndrome de Zellweger; la adrenoleucodistrofia neonatal; y la enfermedad de Refsum infantil
- Los que resultan de múltiples deficiencias enzimáticas (condrodisplasia rizomélica punctata)
- Los que resultan de una sola deficiencia enzimática: adrenoleucodistrofia ligada al cromosoma X e hiperoxaluria primaria tipo 1
El Síndrome de Zellweger, el más letal de los tres trastornos biogénesis peroxisomal, causa hipotonía infantil, convulsiones y la muerte en el primer año. Las manifestaciones oftálmicas incluyen opacificación corneal, catarata, glaucoma, RP y atrofia óptica. La Adrenoleucodistrofia neonatal y la enfermedad infantil de Refsum parecen ser genéticamente distintos, pero clínica, bioquímica, y patológicamente similares al síndrome de Zellweger, aunque más leves.
La Condrodisplasia rizomélica punctata, un trastorno peroxisomal que resulta de por lo menos dos deficiencias enzimáticas peroxisomales, se presenta al nacer con anormalidades esqueléticas; los pacientes rara vez sobreviven más allá de un año de edad y la manifestación ocular más prominente consta de cataratas bilaterales.
La Adrenoleucodistrofia XL resulta de una deficiencia de una sola enzima peroxisomal, se presenta al final de la primera década con deterioro conductual, cognitivo y visual. La pérdida de la visión resulta de desmielinización de toda la vía visual, y la retina externa está preservada. Mientras tanto, la hiperoxaluria primaria tipo 1 se manifiesta por la proliferación de pigmento subretiniano parafoveal.
La identificación precoz de estos trastornos, que pueden depender de reconocer los hallazgos oftalmológicos, es fundamental para el diagnóstico prenatal, tratamiento y asesoramiento genético.148,149
Se inicia en el periodo neonatal, como resultado tanto de la malformación de órganos como de la disfunción peroxisomal que causa un deterioro progresivo.
Los recién nacidos presentan rasgos craneofaciales dismórficos (facies aplanadas, fontanela anterior grande, suturas abiertas, frente alta y prominente, occipucio aplanado, fisuras palpebrales inclinadas hacia arriba, pliegues del epicanto, puente nasal ancho), hipotonía profunda y convulsiones. Pueden presentarse rasgos craneofaciales dismórficos como macrocefalia o microcefalia, paladar ojival, micrognatia y pliegues de piel redundantes en el cuello. Son frecuentes las anomalías esqueléticas (condrodisplasia punctata) y los quistes renales subcorticales. A menudo hay retraso en el crecimiento, hepatomegalia, ictericia y coagulopatía. Los hallazgos oculares incluyen cataratas, glaucoma, RP, nistagmus, opacidad corneal y atrofia del nervio óptico. Los cambios y pérdida visuales son progresivos. Puede presentarse pérdida de audición neurosensorial. Puede producirse criptorquidia e hipospadias y clitoromegalia. La función del sistema nervioso central está gravemente afectada y sufren un retraso psicomotor profundo.134,148
muy baja
Esta enfermedad es completamente diferente del Refsum infantil. Es un trastorno hereditario en el que el ácido fitánico se acumula en los tejidos y suero. Presenta RP progresiva, sordera, alteraciones del movimiento y del equilibrio, alteraciones gustativas y olfativas, neuropatía sensoriomotora, ictiosis, arritmias cardiacas y hasta un 35% de los pacientes presentan metacarpianos o metatarsianos cortos. Para el diagnóstico precoz en estos casos nos apoyaremos en analíticas de sangre que muestren esos niveles elevados de ácido fitánico.134,150 Tenemos que estar especialmente atentos al diagnóstico de esta enfermedad pues puede ser tratada con una dieta pobre en ácido fitánico.151
muy baja
Hipo/abetalipoproteinemias o enfermedad de Bassen-Kornzweig
Se refiere a un grupo de trastornos hereditarios AR de las lipoproteínas caracterizados por bajas concentraciones o ausencia de colesterol, de lipoproteínas de baja densidad y apolipoproteína B en el plasma. La abetalipoproteinemia y la hipobetalipoproteinemia familiar homocigótica, aunque causadas por mutaciones en genes diferentes, son clínicamente indistinguibles.152 Recientemente se ha propuesto un marco para el seguimiento clínico y la gestión de estos dos trastornos, centrándose en la vigilancia del crecimiento en los niños y la prevención de complicaciones, proporcionando asesoramiento dietético especializado aportando altas dosis de vitaminas liposolubles, otros suplementos como el ácido linoleico y los ácidos omega 6, así como regímenes alimenticios que reduzcan la ingesta de grasas. Por lo tanto, tenemos que estar especialmente atentos al diagnóstico de esta enfermedad pues puede ser tratada con vitamina A y vitamina E y, en algunos casos, con la terapia de la vitamina K.151
Clínicamente se caracterizan por niveles indetectables o muy bajos de colesterol LDL y apoB, retraso en el desarrollo, la incapacidad de absorber la grasa de la dieta, diarrea, esteatorrea y acantocitosis, así como la estenosis hepática y de lípidos en los enterocitos.152-154 El déficit de vitaminas liposolubles origina alteraciones neurológicas como la ataxia espinocerebelosa y otras como la RP. Cursa con ataxia, oftalmoplejia, alteración del equilibrio y debilidad muscular.134 A nivel retiniano se ha descrito atrofia macular,155 distrofia retiniana helicoidal peripapilar,156 o estrías angioides,157 pero lo habitual es que se muestre como una pseudo-RP. En el ERG se muestra alteración de respuestas fotópicas y escotópicas que se recuperan tras el tratamiento con Vitamina A.158
muy baja
Degeneración pigmentaria retiniana asociada a nefritis crónica hereditaria y sordera neurosensorial de tonos agudos. Puede asociar microesferofaquia, lenticono anterior, catarata subcapsular posterior, distrofia corneal polimorfa posterior y a veces retinosquisis periférica. Por último, típicamente muestran lesiones amarillentas-blanquecinas puntiformes localizadas tanto en la retina superficial pericentral en área macular como localizadas profundas a los vasos retinianos en la periferia media. Se transmite frecuentemente de forma XL (mutaciones en COL4A3 y COL4A5) aunque se han descrito otros patrones de herencia, como la AD, AR o digénica.159-162
muy baja
Se caracteriza por ser una enfermedad neurocutánea AR que cursa con ictiosis congénita, discapacidad intelectual de leve, paresia espástica simétrica con afectación sobre todo de las piernas, convulsiones, displasia ósea y dental, disminución de la sudoración, hipertelorismo, reducción de la esperanza de vida, y cambios oftalmoscópicos en la mácula caracterizados por la presencia de depósitos blanquecinos brillantes que son los más característicos aunque pueden observarse también cambios pigmentarios amarillos que pueden parecerse a los de la enfermedad de Best.163,164
muy baja
Las lesiones típicas son espasmos en la infancia, agenesia del cuerpo calloso y defectos lacunares coriorretinianos, que puede asociarse con otras anomalías oculares que incluyen microftalmía, colobomas o aplasias del nervio óptico, membrana pupilar persistente y tejido glial que se extiende desde el nervio óptico.165-167
muy baja
Enfermedad AR que se caracteriza por paraparesia espástica y demencia. Algunos de estos pacientes pueden presentar distrofia en patrón.168-171
baja
Síndrome de Joubert
El síndrome de Joubert es una enfermedad AR caracterizada por una malformación peculiar del tronco cerebral y el cerebelo, conocida como el «signo del diente molar». La presentación neurológica de esta enfermedad incluye hipotonía que desemboca en la ataxia y blefaroptosis, retraso en el desarrollo, movimientos oculares anormales y alteraciones respiratorias neonatales. Se asocia a menudo con afectación multiorgánica variables, principalmente de la retina (que va desde la severa ACL a retinopatías lentamente progresivas con visión parcialmente conservada), los riñones y el hígado.
Existe un grupo de enfermedades llamado síndrome de Joubert y trastornos relacionados (JSRD), que comparten una afectación multiorgánica. Hasta la fecha se han identificado 29 genes causales, todos ellos codifican proteínas expresadas en el cilio primario o su aparato.172 De hecho, JSRD presenta solapamiento clínico y genético con un campo cada vez mayor de trastornos debido a mutaciones en proteínas ciliares, que se conocen colectivamente como «ciliopatías».173 Estos incluyen el síndrome Senior-Loken y el síndrome de Bardet-Biedl entre los que pueden presentar afectación retiniana. En este sentido se han descrito correlaciones significativas genotipo-fenotipo entre estas presentaciones clínicas y mutaciones específicas en los genes JSRD, con implicaciones relevantes en términos de diagnóstico molecular, seguimiento clínico y tratamiento de los pacientes que presentan estos cuadros. Por otra parte, la identificación de mutaciones permite el diagnóstico prenatal temprano en parejas en situación de riesgo, mientras que la neuroimagen fetal puede no ser informativa hasta finales del segundo trimestre del embarazo.174-176
muy baja
Síndrome de Senior-Loken
Ciliopatía que produce degeneración pigmentaria tapetorretiniana y nefronoptisis.137
muy baja
Síndrome de Alström
Es otra ciliopatía AR que cursa con degeneración pigmentaria retiniana, sordera neurosensorial, diabetes mellitus tipo 2 y obesidad. Los primeros síntomas aparecen en la infancia, con una gran variabilidad en cuanto a la gravedad y la evolución clínica incluso dentro de la misma familia.134,137,177
muy baja
| Calidad baja/muy baja |
En las DHR sindrómicas aparecen signos y síntomas oculares similares a los que se observan en los casos aislados de DHR.90,91 |
| Calidad muy baja |
En las DHR sindrómicas los hallazgos oftalmológicos pueden ser sutiles y/o aparecer a posteriori.92 |
| Calidad muy baja |
En las DHR sindrómicas no todos los signos y síntomas sistémicos aparecen en todos los pacientes.131 |
| Calidad muy baja |
Varias DHR asocian sordera y degeneración retiniana, siendo el USH el más frecuente.92,134 |
| Calidad muy baja |
El síndrome de Bardet-Biedl es la 2ª DHR sindrómica en frecuencia. Los signos y síntomas oculares y sistémicos aparecen en la adolescencia. Asocia obesidad troncular, hipogonadismo, retraso mental, polidactilia y/o braquisindactilia entre otros.90,138-140 |
| Calidad muy baja |
Algunas DHR sindrómicas como el síndrome de Lawrence-Moon, USH, Refsum, y las Hipo/abetalipoproteinemias, pueden asociar trastornos del movimiento, del equilibrio y/o de la marcha entre otros.134,137,138 |
| Calidad muy baja |
Algunas DHR sindrómicas como el síndrome de Senior-Loken, el síndrome de Bardet-Biedl y el síndrome Cerebro-Hepato-Renal pueden asociar alteraciones renales.90,134,137 |
| Calidad muy baja |
El Síndrome Kearns Sayre se asocia a distintas formas de degeneración retiniana (se ha descrito RP típica y también atrofia similar a la que produce la distrofia coroidea areolar central), oftalmoplejia externa progresiva, fatiga muscular, insuficiencia mitral alteraciones en el electrocardiograma (bloqueo auriculoventricular),137,142 diabetes,143 dismorfias faciales y/o retraso psicomotor.134,137,141 |
| Calidad muy baja |
La Distrofia miotónica de Steinert o miotonía atrófica se caracteriza por miotonía, con atrofia muscular selectiva, calvicie, atrofia testicular y ovárica, senilidad prematura y cataratas. Un 20-25% de estos pacientes muestra evidencia de degeneración retiniana.144 Las complicaciones oculares en la distrofia miotónica incluyen ptosis, debilidad de los músculos oculares, cataratas, respuesta pupilar lenta, presión intraocular baja y depósitos amarillentos trirradiados o pigmentados de configuración pavimentosa en la periferia de la retina.145 |
| Calidad muy baja |
La distrofia muscular de Duchenne se caracteriza por debilidad muscular proximal progresiva. El 95% de los pacientes habrá perdido la capacidad para caminar a la edad de 12 años y la mortalidad es del 95% a los 20 años. La mayor parte de los pacientes tienen ERG con amplitudes subnormales, hiperpigmentación focal de la mácula; pero son normales la AV, la visión de color y la función de los músculos extraoculares.146 |
| Calidad muy baja |
El síndrome de Zellweger causa hipotonía infantil, convulsiones y la muerte en el primer año.148,149 Los recién nacidos presentan rasgos craneofaciales dismórficos (facies aplanadas, fontanela anterior grande, suturas abiertas, frente alta y prominente, occipucio aplanado, fisuras palpebrales inclinadas hacia arriba, pliegues del epicanto, puente nasal ancho). Son frecuentes las anomalías esqueléticas (condrodisplasia punctata) y los quistes renales subcorticales. A menudo hay retraso en el crecimiento, hepatomegalia, ictericia y coagulopatía. Las manifestaciones oftálmicas incluyen opacificación corneal, catarata, glaucoma, RP y atrofia óptica. Los cambios y pérdidas visuales son progresivos. Puede presentarse pérdida de audición neurosensorial.134,148,149 |
| Calidad muy baja |
El síndrome Refsum del adulto presenta RP progresiva, sordera, alteraciones del movimiento y del equilibrio, alteraciones gustativas y olfativas. Para el diagnóstico precoz se realizará una analíticas de sangre sobre los niveles elevados de ácido fitánico.134,150 |
| Calidad muy baja |
La Hipo/abetalipoproteinemias o Enfermedad de Bassen-Kornzweig se caracteriza por bajas concentraciones, o ausencia de colesterol, de lipoproteínas de baja densidad, apolipoproteína B en el plasma,152 retraso en el desarrollo, la intolerancia a la grasa alimenticia, diarrea, esteatorrea y acantocitosis, así como la estenosis hepática y de lípidos en los enterocitos.152-154 Cursa con ataxia, oftalmoplejia, alteración del equilibrio y debilidad muscular.134 A nivel retiniano se ha descrito atrofia macular,155 distrofia retiniana helicoidal peripapilar,156 o estrías angioides,157 pero lo habitual es que se muestre como una pseudo-RP. Puede ser tratada con vitamina A y vitamina E y, en algunos casos, con la terapia de la vitamina K.151 En el ERG se muestra alteración de respuestas fotópicas y escotópicas que se recuperan tras el tratamiento con Vitamina A.158 |
| Calidad muy baja |
El síndrome de Alport es una degeneración pigmentaria retiniana asociada a nefritis crónica hereditaria y sordera neurosensorial de tonos agudos. Puede asociar microesferofaquia, lenticono anterior, catarata subcapsular posterior, distrofia corneal polimorfa posterior y a veces retinosquisis periférica. Muestran lesiones amarillentas-blanqueadas puntiformes localizadas tanto en la retina superficial pericentral en área macular como localizadas profundas a los vasos retinianos en la periferia media.159-162 |
| Calidad muy baja |
El síndrome de Sjögren-Larsson se caracteriza por ictiosis congénita, discapacidad intelectual de bajo grado, paresia espástica simétrica con afectación sobre todo de las piernas, convulsiones, displasia ósea y dental, sudoración defectuosa, hipertelorismo, reducción de la esperanza de vida, y cambios oftalmoscópicos en la mácula caracterizados por la presencia de depósitos blanquecinos brillantes.163,164 |
| Calidad muy baja |
El síndrome de Aicardi se caracteriza por espasmos en la infancia, agenesia del cuerpo calloso y defectos lacunares coriorretinianos, que puede asociarse con otras anomalías oculares que incluyen microftalmía, colobomas o aplasias del nervio óptico, membrana pupilar persistente y tejido glial que se extiende desde el nervio óptico.165-167 |
| Calidad muy baja |
El síndrome de Kjellin se caracteriza por paraparesia espástica, demencia y, en ocasiones, de distrofia en Patrón.168-171 |
| Calidad muy baja |
El síndrome de Joubert se caracteriza por una malformación peculiar del tronco cerebral y el cerebelo, conocida como el «signo del diente molar». La presentación neurológica de esta enfermedad incluye hipotonía que desemboca en la ataxia, retraso en el desarrollo, movimientos oculares anormales y alteraciones respiratorias neonatales. Se asocia a menudo con afectación multiorgánica variables, principalmente de la retina, los riñones y el hígado.174-176 La afectación retiniana varía desde una retinopatía lentamente progresiva con visión parcialmente conservada hasta la severa ACL. |
| Calidad muy baja |
El síndrome de Senior-Loken produce RP y nefronoptisis.137 |
| Calidad muy baja |
El síndrome de Alstrom cursa con degeneración pigmentaria retiniana, sordera neurosensorial, diabetes mellitus tipo 2 y obesidad.134,137,177 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- Calidad global de la evidencia: la calidad de los estudios seleccionados fue baja o muy baja, lo que determinó la fuerza de la recomendación.
- Costes y uso de recursos: No se han identificado estudios que valoren los costes asociados.
El grupo elaborador formuló las siguientes recomendaciones considerando qué signos y síntomas de sospecha presentan las DHR sindrómicas.
Recomendaciones
| Condicional | Se sugiere sospechar de síndrome de Usher en niños con sordera neurosensorial profunda y retraso de la marcha independiente. |
| Condicional | Se sugiere estudiar precozmente a todos los neonatos con sordera preverbal neurosensorial severa y profunda a través de una exploración clínica que incluya test electroneurofisiológicos auditivos, vestibulares y oftalmológicos. |
| Condicional | Se sugiere no excluir definitivamente el diagnóstico de cualquier DHR sindrómica por la ausencia de signos oftalmoscópicos evidentes en la exploración inicial. |
| Condicional | Ante la sospecha de cualquier DHR sindrómica en ausencia de signos oftalmoscópicos evidentes en la exploración, se sugiere el test de electrofisiología para su diagnóstico. |
5.2.5. Estudio genético
- ¿Cuándo está indicado realizar un estudio genético en personas con DHR? ¿Cómo se debe realizar?
El planteamiento de los estudios diagnósticos genéticos ha variado radicalmente a partir de la aparición de la NGS en la práctica clínica,178 con su potente capacidad para identificar mutaciones y causas genéticas en enfermedades genéticamente heterogéneas al permitir secuenciar de un modo razonablemente rápido y a un coste asequible un elevado número de genes. Aunque estos estudios plantean otros problemas, tales como el almacenamiento de los datos, el análisis bioinformático de las secuencias, la correcta interpretación de los resultados obtenidos (patogenicidad de las variantes versus asociaciones espurias), la aparición de hallazgos inesperados y el abordaje ético legal de todo ello, entre otras. Pero, sin duda, el conocimiento generado y su aplicación clínica han supuesto un avance en el abordaje diagnóstico de las DHR.
Mientras en la etapa anterior a la NGS, el análisis de los genes se realizaba uno a uno, con el consiguiente retraso diagnóstico, a veces de años y con un pobre rendimiento diagnóstico (aproximadamente 25% de los casos).179 Actualmente, el diagnóstico en el caso de paneles de genes se demora solo unas semanas y la técnica tiene un rendimiento cercano al 50% de casos resueltos. Este cambio hace que, en la revisión sistemática realizada para dar respuesta a esta pregunta clínica, el planteamiento sea muy diferente dependiendo de la fecha de la publicación.
Aun así y aunque Heon et al. en 2003180 plantea numerosos interrogantes: ¿Cómo organizar las pruebas de ácido desoxirribonucleico (ADN)? ¿Qué genes analizar? ¿Dónde hacerse? ¿Cuánto costaría? ¿Cómo de exactos son los resultados? Y ¿cómo va a cambiar el manejo del paciente?; MacDonald et al. en 2004181 informaron que la capacidad de proporcionar diagnósticos genéticos exactos ha permitido el asesoramiento adecuado a los pacientes y las familias.
La respuesta a esta pregunta clínica se basa, por un lado, en la Orden SSI/2065/2014, de cartera de servicios comunes del SNS, que plantea un marco general para la aplicación de los estudios genéticos diagnósticos182 y, por otro, en 55 documentos identificados que recogen opiniones de expertos (n=30) y estudios observacionales (n=25) en pacientes con diferentes DHR entre las que se encuentran la RP, coroideremia, el USH, las distrofias de conos y bastones, la enfermedad de Stargardt, la ACL, la vitreorretinopatía exudativa familiar y el síndrome de Bardet-Biedl. Para conocer el listado de los genes implicados en DHR consultar RetNet (https://sph.uth.edu/retnet/)
La Orden SSI/2065/2014, de cartera de servicios comunes del SNS recoge los distintos tipos y situaciones en la que puede hacerse un diagnóstico genético, incluyendo los análisis genéticos diagnósticos, presintomáticos, de portadores, prenatal y preimplantacional.182
Los distintos autores recogen igualmente todas estas situaciones, en las que recomiendan su aplicación:
muy baja
Es importante resaltar que, de acuerdo a la citada Orden SSI/2065/2014, las recomendaciones para realizar un estudio presintomático incluyen que la persona pertenezca a una familia o grupo poblacional de alto riesgo en los que se ha identificado la presencia de una enfermedad o trastorno genético y que además, el estudio permita o bien diagnóstico precoz y un tratamiento más temprano o la toma de decisiones reproductivas del individuo o de sus familiares que pueden comprometer a su descendencia. Por otra parte, en el caso de menores, el estudio genético presintomático en enfermedades que aparecen en la edad adulta se deberá diferir hasta que la persona tenga la madurez y competencia necesaria para comprender la naturaleza e implicaciones de su decisión, salvo que existan medidas preventivas eficaces aplicables en la infancia. En enfermedades que aparecen en la infancia y que pueden ser prevenidas o tratadas adecuadamente se deberá realizar lo más cercano posible a la fecha en la que se deben iniciar dichas medidas preventivas y/o terapéuticas.182
En cuanto a las condiciones clínicas en las que realizar el diagnóstico genético, en la bibliografía se recogen un gran número de patologías, englobadas todas genéricamente bajo el epígrafe de distrofias de retina,196-198 entre las que se incluyen: formas no sindrómicas de RP,199 RP de inicio precoz y afectación macular,200 ceguera nocturna congénita estable199, coroideremia32, enfermedad de Best,199 enfermedad de Stargardt,201 distrofia de conos y bastones,202 patologías debidas a mutaciones en ABCA4,203 y vitreorretinopatías,204 así como formas sindrómicas de RP205 tales como el USH,206,207 el síndrome de Bardet-Biedl,208 otras DHR asociadas a enfermedad renal entre otras,209 y ACL sindrómica o no.210-213
baja/muy
baja
Con respecto a cuándo está indicado realizar un estudio genético, la Orden SSI/2065/2014, de cartera de servicios comunes del SNS de los estudios genéticos diagnósticos,182 establece que los análisis genéticos incluidos en la cartera común de servicios del SNS deben cumplir los siguientes requisitos:
- Tener validez analítica y clínica sustentada en la evidencia científica
- Ser de utilidad clínica: Constituir un elemento esencial para el diagnóstico, pronóstico, selección y seguimiento de tratamientos, así como para tomar decisiones reproductivas, siempre que el balance beneficio/riesgo sea favorable
- Haber sido valorados previamente en relación a las implicaciones éticas, sociales, legales, organizativas y económicas de su inclusión en la oferta asistencial pública
Encontrar las mutaciones causantes de las DHR puede ser difícil debido al gran número de genes responsables, así como a la complejidad de la relación genotipo fenotipo en estos trastornos. Sin embargo, un diagnóstico molecular preciso es fundamental, no sólo para la terapia genética, sino también para el asesoramiento genético, la prescripción de fármacos específicos de la ruta,210 y el establecimiento del pronóstico de la enfermedad.
El diagnóstico molecular actualmente forma parte del diagnóstico integral del paciente,210 al refinar el diagnóstico clínico en casos de ambigüedad fenotípica.210,214,215
Estos estudios además están aclarando la etiopatogenia y redefiniendo el espectro clínico de las DHR, explicando el solapamiento observado en fenotipos clínicos y anticipando otros problemas médicos oculares o sistémicos.188
Además permiten realizar una estimación pronóstica más precisa,214,215 y el asesoramiento genético al paciente y su familia, produciendo una mejora en su atención y favorece la esperanza en posibles tratamientos mediante terapia génica, tal y como parecen indicar algunos ensayos clínicos en ACL.214,215
baja/muy
baja
Solo un artículo, anterior a la era de los ensayos clínicos en humanos y de la NGS cuestiona esta utilidad.216
Los distintos autores detallan en qué consiste la llamada «utilidad clínica», esto es cuando podría existir alguna mejora en el manejo del paciente (presente o futuro) o beneficios para sus familiares.
Esta mejora en el manejo clínico se puede resumir en los siguientes aspectos:
- Confirmación, mejora (refinamiento) o reclasificación del diagnóstico clínico187,188,192,198,210,214,215,217-219
- Anticipación de otros problemas médicos188 (formas sindrómicas) o su descarte
- Valor pronóstico118,188,215,220
- Deseo de conocimiento por el paciente: bienestar psicológico178
- Asesoramiento genético a la familia181,188,192,198,214,215,221
- Opciones futuras terapéuticas188,198,210,212-215,220
- Medidas preventivas. Evitar la vitamina A en caso mutación en ABCA4
baja/muy
baja
Con respecto a cómo realizar un estudio genético, la Orden SSI/2065/2014 establece que la atención a los pacientes y familiares en el área de genética en el SNS incluirá:
a) El diagnóstico de enfermedades o trastornos de base genética, mediante la integración de la información clínica personal y familiar y la obtenida tras la realización de los estudios genéticos.
b) La transmisión de información, de forma clara y comprensible, sobre el riesgo de recurrencia de la enfermedad o trastorno, las consecuencias para el paciente y su descendencia y las posibilidades de prevención pre- y post-natal.
c) La derivación de los pacientes y familiares a los distintos profesionales especializados y grupos de apoyo necesarios para el adecuado manejo de cada situación.
Además, establece que el proceso de asesoramiento genético y de realización de análisis genéticos con fines sanitarios deberá ser efectuado por personal cualificado y deberá llevarse a cabo en centros acreditados que reúnan los requisitos de calidad que reglamentariamente se establezcan al efecto, tal como establece el artículo 56 de la Ley 14/2007, de 3 de julio, de Investigación biomédica. Asimismo, la autoridad autonómica o estatal competente acreditará los centros, públicos o privados, que puedan realizar análisis genéticos.
Distintos autores detallan cómo realizar el diagnóstico genético enumerando alguno de los procesos implicados. Así, se resalta la necesidad de ofrecer estas pruebas a los pacientes cuando estén indicadas, usando el árbol genealógico como primer paso190 y el apoyo de un fenotipado preciso para orientar el estudio.209
Varias publicaciones coinciden en la pertinencia de asociar el asesoramiento genético pre- y post-test a la práctica de estudios genéticos diagnósticos en DHR,192,193,222 así como en la necesidad de requerir el consentimiento informado, que en España es obligatorio de acuerdo con la LIB 14/2007.222 Igualmente se deben tomar en consideración aspectos tales como el coste, el tiempo de demora del diagnóstico, el tipo de informe y la utilidad clínica.178
muy baja
Por todo lo anterior, en los últimos años, las publicaciones reflejan el cambio experimentado en la aproximación al diagnóstico genético, anteriores223,224 o tras la implementación de la NGS en la práctica clínica, coincidiendo todos los autores en que ésta debe ser la aproximación de elección (paneles de genes)219,225 para los casos de heterogeneidad genética,188,196-198,201,203,210213,215,225-228 quedando el estudio de secuenciación convencional de un solo gen para condiciones nada o poco heterogéneas (tales como coroideremia, retinosquisis)179 que afectan a genes de tamaño pequeño o mediano y los estudios de secuenciación de exoma o genoma completo para aquellos casos especiales y no resueltos mediantes la NGS de paneles y más bien en el ámbito de la investigación.
Resumen de la evidencia
| Calidad baja / muy baja |
Entre las condiciones clínicas en las que realizar el diagnóstico se incluyen: formas no sindrómicas de RP,199 RP de inicio precoz y afectación macular,200 ceguera nocturna congénita estable,199 coroideremia,32 enfermedad de Best,199 enfermedad de Stargardt,201 distrofia de conos y bastones,202 patologías debidas a mutaciones en ABCA4,203 y vitreorretinopatías hereditarias.204 |
| Calidad baja / muy baja |
Asimismo, se incluyen formas sindrómicas de RP205 tales como el USH,206,207 el síndrome de Bardet-Biedl,208 otras DHR asociadas a enfermedad renal entre otras,209 y ACL sindrómica o no.210-213 |
| Calidad baja / muy baja |
El diagnóstico genético actualmente forma parte del diagnóstico integral del paciente,210 refina el diagnóstico clínico,210,214,215aclara la etiopatogenia y redefine el espectro clínico de las DHR, anticipando otros problemas médicos oculares o sistémicos,188 permite realizar una estimación pronóstica precisa y el asesoramiento genético al paciente y su familia.214,215 |
| Calidad baja / muy baja |
La utilidad clínica del diagnóstico genético se puede resumir en los siguientes aspectos: confirmación, mejora (refinamiento) o reclasificación del diagnóstico clínico,187,188,192,210,214,215,217-219 anticipación de otros problemas médicos,188 valor pronóstico,118,188,215,220 cobertura del deseo de conocimiento por el paciente: bienestar psicológico,178 asesoramiento genético a la familia,181,188,192,198,214,215,221 opciones futuras terapéuticas188,198,210,212-215,220 y medidas preventivas. |
| Calidad muy baja |
El diagnóstico genético se puede realizar usando el árbol genealógico como primer paso190 y para el apoyo de un fenotipado preciso para orientar el estudio.209 |
| Calidad muy baja |
Asociar el asesoramiento genético previo y posterior a la práctica de estudios genéticos diagnósticos en DHR parece pertinente.192,193,222 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- Calidad global de la evidencia: la evidencia disponible deriva de diversas cohortes, series de casos y revisiones de casos aislados, considerándose la calidad global de la evidencia como muy baja.
- Valores y preferencias de los pacientes: se identificaron varios estudios que exploran los valores y preferencias de pacientes con DHR y sus familiares sobre las pruebas genéticas. Los principales resultados se resumen a continuación:
En el estudio llevado a cabo en Reino Unido por Willis et al.,229 la mayoría (90%) consideró la prueba buena o muy buena y que se sometería a ella si se la ofrecieran. Además, el 87% consideraba que la prueba debe ser ofrecida sólo después de que el individuo ha recibido asesoramiento genético de un profesional.
En esta misma línea, los encuestados en Bong et al. consideraron que las pruebas genéticas deben estar disponible para todos los afectados (92%) y también para los familiares no afectados (78%), además, la mayoría afirmó que la prueba genética debería estar disponible sólo después del asesoramiento genético (72%). Las principales razones dadas a favor de la prueba fueron: identificar un nuevo tratamiento (89%), ayudar en la comprensión de la causa de la discapacidad visual (81%), confirmar la forma en que se hereda la condición (78%), y predecir el futuro función visual (75%).230
En Combs et al.,184 los entrevistados, tanto afectados como no afectados, pertenecientes a familias con DHR de Reino Unido, expresaron puntos de vista principalmente positivos sobre la prueba genética, destacando beneficios tales como la confirmación del diagnóstico, la información sobre los riesgos y una preparación mejor para el futuro.184 En la misma línea, en otro estudio realizado en los Estados Unidos de América con pacientes con USH, estos informaron de que las pruebas genéticas tienen importantes implicaciones positivas para la seguridad de la infancia, la planificación de la educación, el asesoramiento genético y el tratamiento.207
En otro estudio realizado en Reino Unido por McKibbin et al.207 los pacientes manifestaron que la información derivada de las pruebas genéticas deberían darse en una consulta cara a cara y acompañada de un formato accesible para las personas con discapacidad.231
En el estudio cualitativo con pacientes con DHR en el contexto español, los pacientes confirmaron los valores y preferencias ya señalados en la literatura. - Costes y uso de recursos: El coste actual de la NGS oscila entre los 500 y los 1500€, incluidos los costes de personal y de amortización de los equipos, para estudios con paneles de genes. La aparición de la tecnología, su potente capacidad para identificar mutaciones y causas genéticas en enfermedades genéticamente heterogéneas, permite secuenciar de un modo razonablemente rápido y a un coste moderado un elevado número de genes. Las actuales técnicas de NGS permiten caracterizar un elevado porcentaje de casos (alrededor del 50%) incluso sin tener un diagnóstico clínico previo muy preciso y ayudar al diagnóstico oftalmológico y al asesoramiento genetico.
- Accesibilidad: Existe un desigual reparto de los servicios de genética dotados de la tecnología NGS disponible para el estudio de las DHR en el territorio nacional.
El grupo elaborador formuló las siguientes recomendaciones sobre las pruebas genéticas en las DHR.
Recomendaciones
| Condicional | Se sugiere realizar un estudio genético ante la sospecha de formas no sindrómicas de RP, RP de inicio precoz y afectación macular, ceguera nocturna congénita estable, coroideremia, enfermedad de Best, enfermedad de Stargardt, distrofia de conos y bastones, patologías debidas a mutaciones en ABCA4, y vitreorretinopatías hereditarias. |
| Condicional | Se sugiere realizar un estudio genético ante la sospecha de formas sindrómicas de RP tales como el síndrome de Usher, el síndrome de Bardet- Biedl o la amaurosis congénita de Leber. |
| Condicional | Se sugiere valorar individualmente la realización de diagnóstico genético en niños. |
| Condicional | Se sugiere valorar individualmente la realización de diagnóstico genético dentro de la planificación familiar. |
| Condicional | Se sugiere el diagnóstico molecular para la terapia genética, el asesoramiento genético, la prescripción de fármacos específicos de la ruta y el establecimiento del pronóstico de la enfermedad. |
| Condicional | Se sugiere asociar el asesoramiento genético previo y posterior a la práctica de estudios genéticos diagnósticos en DHR. |
5.3. Asesoramiento genético
- ¿Cuándo está indicado el asesoramiento genético en personas con DHR? ¿cómo se debe realizar?
El asesoramiento genético es un proceso médico por el cual se informa y apoya a aquellas personas y familias que tienen una enfermedad hereditaria o riesgo de tenerla, brindándoles información acerca de la enfermedad, el riesgo de transmitirlo a su descendencia y las medidas de manejo médico y psicológico que se les puede ofrecer, ayudándoles a tomar decisiones informadas.
La respuesta a estas preguntas clínicas se basa, por un lado, en la Orden SSI/2065/2014, de Cartera de Servicios Comunes del SNS182 y la Ley 14/2007, de 3 de julio, de Investigación Biomédica (LIB)232 y, por otro, en 21 documentos identificados que recogen opiniones de expertos (n=7) y estudios observacionales (n=14) en pacientes con diferentes DHR entre las que se encuentran la RP, coroideremia, el USH, las distrofias de conos y bastones, la ACL, la vitreorretinopatía exudativa familiar y la distrofia macular de Sorby.
Como en el caso del diagnóstico genético, en España, el asesoramiento genético está definido por las mencionadas Ley de Investigación biomédica232 y la Orden de Cartera de Servicios Comunes del SNS.182 En ambos documentos se define como «el procedimiento destinado a informar a una persona sobre las posibles consecuencias para él o su descendencia de los resultados de un análisis o cribado genético y sus ventajas y riesgos y, en su caso, para asesorarla en relación con las posibles alternativas derivadas del análisis. Este procedimiento tendrá lugar tanto antes como después de una prueba o cribados genéticos e incluso en ausencia de los mismos» (para más información sobre diagnóstico genético, ver pregunta 5).
La cartera de servicios regula el asesoramiento genético de modo que lo considera como parte de la atención a familiares y pacientes en el área de genética, incluyendo «la transmisión de información, de forma clara y comprensible, sobre el riesgo de recurrencia de la enfermedad o trastorno, las consecuencias para el paciente y su descendencia y las posibilidades de prevención pre- y post-natal» (apartado 5.3.10.1.b de la Orden SSI/2065/2014). Asimismo, establece que «deberá ser efectuado por personal cualificado y deberá llevarse a cabo en centros acreditados que reúnan los requisitos de calidad que reglamentariamente se establezcan al efecto» (apartado 5.3.10.2 de la Orden SSI/2065/2014) y marca entre sus objetivos «ayudar a la persona o familia a entender y adaptarse a las consecuencias médicas, psicológicas, familiares y sociales de una determinada enfermedad o trastorno genético» (apartado 5.3.10.3.a de la Orden SSI/2065/2014).
Así mismo, la cartera de servicios indica cómo debe llevarse a cabo este asesoramiento (apartado 5.3.10.3.a de la Orden SSI/2065/2014). Este proceso, que incluye la intervención de uno o más profesionales, consistirá en:
1º. Interpretar los antecedentes médicos personales o familiares que permiten valorar el riesgo de ocurrencia o recurrencia de una enfermedad o trastorno de base genética.
2º. Informar sobre el tipo de herencia, los análisis genéticos y sus consecuencias, la posibilidad de prevención o tratamiento y la disponibilidad y accesibilidad de recursos.
3º. Ofrecer el apropiado asesoramiento, respetando el principio de autonomía de las personas para la toma de decisiones.
4º. Solicitar el consentimiento informado previamente a la realización de cualquier análisis genético, tras explicar su validez y utilidad clínica, sus beneficios y las consecuencias derivadas de realizarlo.
La correcta interpretación de los estudios diagnósticos genéticos permite obtener un mejor conocimiento de los riesgos personales y familiares y la toma de decisiones reproductivas.180184
El asesoramiento genético resulta imprescindible, no solo después de la realización de las pruebas genéticas, sino previamente a éstas, durante el proceso de consentimiento informado, dadas las implicaciones psicosociales que pueden tener las enfermedades genéticas.222 El asesoramiento genético permite anticipar los distintos posibles resultados, incluidos los inciertos, los hallazgos inesperados, la ausencia de resultados o las mutaciones genéticas predictivas, para que así el paciente pueda optar por realizarse o no los estudios o elegir qué resultados conocer.
muy baja
Sobre el cuándo se debe indicar el asesoramiento genético, la Orden SSI/2065/2014 (apartado 5.3.10.3.b) establece que, al menos, ante el diagnóstico, sospecha diagnóstica o antecedentes familiares de diversas situaciones que incluyen entre otras las enfermedades hereditarias infantiles y del adulto y las anomalías congénitas y del desarrollo.
Varios autores describen las situaciones que requieren el asesoramiento genético. Tal y como se dice más arriba, deberá realizarse antes y después de una prueba genética,181,222 cuando se realiza el diagnóstico clínico207,233 y se sienta la indicación de un estudio para señalar sus riesgos y beneficios, el tipo, cantidad y fiabilidad de la información que se podría obtener, así como la posibilidad de hallazgos inesperados.189
De modo particular, en el caso de diagnóstico prenatal195,234, o de sujetos asintomáticos, acompañando a un test predictivo o a un estudio de portador.180,186,192,235
muy baja
Es importante resaltar que el asesoramiento genético, esto es la información sobre los riesgos personales y reproductivos que permita tomar decisiones terapéuticas210 y reproductivas198,217 y que debe proporcionarse a toda persona con una enfermedad genética,221 incluidos casos de pacientes pediátricos con síntomas,204 puede iniciarse incluso en ausencia de una prueba genética.222 Sin embargo, este escenario, común hasta hace poco en que el diagnóstico genético era poco accesible, en la actualidad debería restringirse a aquellos casos en los que, tras aplicar las pruebas disponibles (a menudo incluyendo NGS mediante paneles de genes), queda sin identificarse su causa genética.
muy baja
Asimismo, en el pasado, aunque los clínicos se daban cuenta de que una prueba genética puede permitir proporcionar asesoramiento más preciso sobre una enfermedad, la mayoría tendía a subestimar el valor de este asesoramiento a menos que hubieran tenido experiencia específica en casos concretos.213 Por el contrario, el asesoramiento genético es percibido por pacientes y familiares de un modo positivo, destacando beneficios tales como la información sobre los riesgos, una preparación mejor para el futuro,184 permitiéndoles elecciones informadas y planificación reproductiva y de sus vidas.236
muy baja
En todos los tipos de distrofia de retina monogénicas está indicado el asesoramiento genético.185,217 Diversos autores lo refieren a propósito de patología concretas, tales como la RP,198 el USH,207,231 la ACL,210 la vitreorretinopatía exudativa familiar204 y la coroideremia.234
muy baja
La información genética es en sí misma compleja de interpretar y de aplicar a nivel clínico, por lo que requiere que el profesional que la transmita tenga un alto nivel de conocimiento y experiencia en la disciplina y en el asesoramiento genético.237
muy baja
Resumen de la evidencia
| Calidad muy baja |
La interpretación de los estudios genéticos facilita el conocimiento de los riesgos personales y familiares y la toma de decisiones reproductivas.180,184 |
| Calidad muy baja |
El asesoramiento genético deberá realizarse antes y después de una prueba genética.181,222 |
| Calidad muy baja |
El asesoramiento genético deberá realizarse cuando se realiza el diagnóstico clínico.207,233 |
| Calidad muy baja |
El asesoramiento genético deberá realizarse en el diagnóstico prenatal.195,234 |
| Calidad muy baja |
El asesoramiento genético deberá realizarse en sujetos asintomáticos, acompañando a un test predictivo o a un estudio de portador.180,186,192,235 |
| Calidad muy baja |
El asesoramiento genético puede iniciarse incluso en ausencia de una prueba genética.222 |
| Calidad muy baja |
La información sobre los riesgos, la mejor preparación para el futuro,184 así como las elecciones informadas y la planificación reproductiva236 se encuentran entre los beneficios percibidos por los pacientes con DHR. |
| Calidad muy baja |
El asesoramiento genético está indicado en todos los tipos de distrofia de retina monogénicas.185,217 |
| Calidad muy baja |
El asesoramiento genético requiere que el profesional que la transmita tenga un alto nivel de conocimiento y experiencia en la disciplina.237 |
De la evidencia a la recomendación
Los aspectos que han determinado la fuerza y la dirección de las recomendaciones formuladas han sido los siguientes:
- Calidad global de la evidencia: la evidencia deriva de diversas opiniones de expertos, cohortes, revisiones de casos y series de casos de baja o muy baja calidad, lo que influyó en la fuerza de la recomendación.
- Costes y uso de recursos: No se han identificado estudios que valoren los costes asociados.
- Valores y preferencias: Se identificaron varios estudios relacionados con los valores y preferencias de pacientes y familiares de pacientes con DHR sobre el asesoramiento. Los principales resultados se resumen a continuación:
En el estudio desarrollado por Willis et al.229 llevado a cabo en Reino Unido, la mayoría (90%) consideró la prueba buena o muy buena y que se sometería a ella si se la ofrecieran. Además, el 87% consideraba que la prueba debe ser ofrecida sólo después de que el individuo ha recibido asesoramiento genético de un profesional. En esta misma línea, en el estudio de Bong et al., los encuestados consideraron que las pruebas genéticas deben estar disponibles para todos los afectados (92%) y también para los familiares no afectados (78%), además, la mayoría afirmó también que la prueba genética debería estar disponible sólo después del asesoramiento genético (72%). Prácticamente la totalidad de los pacientes y los familiares de pacientes del estudio de Mezer et al., realizado en Canadá, están completamente de acuerdo con el hecho de que el individuo debe ser educado y aconsejado sobre la naturaleza de la prueba antes de someterse a esta.
Por último, en el estudio llevado a cabo en Reino Unido por McKibbin et al.207 los pacientes manifestaron que la información derivada de las pruebas genéticas deberían darse en una consulta cara a cara y acompañada de un formato accesible para las personas con discapacidad.231
En el estudio cualitativo con pacientes con DHR en el contexto español, los pacientes confirmaron los valores y preferencias ya señalados en la literatura. Aparte de los beneficios señalados previamente, todas las personas entrevistadas valoraron positivamente la posibilidad de tener información preconcepcional que les permita planificar sus decisiones reproductivas.
Recomendaciones
| Condicional | Se sugiere el asesoramiento genético en el diagnóstico de todos los tipos de DHR monogénicas. |
| Condicional | Se sugiere el asesoramiento genético antes y después del estudio genético. |
| √ | Se sugiere que durante el asesoramiento genético previo al diagnóstico genético se exploren las preocupaciones y expectativas del paciente con respecto a las pruebas genéticas. |
| √ | Se sugiere que durante el asesoramiento genético previo al diagnóstico genético se anticipe la posibilidad de hallazgos negativos, inciertos o inesperados y se dé al paciente la opción de realizar o no la prueba genética. |
| √ | En caso de decidir realizar el diagnóstico genético, se sugiere acordar con el paciente qué tipo de resultados desea conocer. |
| √ | En caso de decidir realizar el diagnóstico genético, se sugiere ofrecer la opción al paciente de que la información derivada de las pruebas genéticas se dé en una consulta cara a cara y acompañada de un formato adecuado a la discapacidad de cada paciente. |
| √ | Se sugiere informar a los pacientes con DHR y a sus familiares de la importancia de recibir asesoramiento genético antes de tomar decisiones reproductivas, así como de la valoración del riesgo de transmisión a la descendencia y las alternativas existentes para evitar la transmisión de la enfermedad. |
| Condicional | Se sugiere que en el asesoramiento genético se facilite información sobre los riesgos personales y familiares. |
| Condicional | Se sugiere que el asesoramiento genético acompañe al diagnóstico clínico. |
| Condicional | Se sugiere el asesoramiento genético en el diagnóstico prenatal. |
| Condicional | Se sugiere el asesoramiento genético en sujetos asintomáticos, acompañando a un test predictivo o a un estudio de portador. |
Bibliografía 5. Diagnóstico
2. Nájera C, Millán J, Beneyto M, et al. Epidemiology of retinitis pigmentosa in the Valencian community (Spain). Genet Epidemiol 1995; 12: 37–46.
3. Fundación Retina España. PREVARET: Estudio Epidemiológico Sobre La Prevalencia De Las Enfermedades Distróficas De La Retina. http://retina.es/retina/index.php/informacion-arm-a-fre/proyectos-cientificos/741-prevaret (2012).
6. Givre S, Gard S. Retinitis pigmentosa: Clinical presentation and diagnosis. UpToDate 2014; 1–12.
18. Coco-Martín R, Navarro R, Pinilla I, et al. Guía clínica para el diagnóstico diferencial y el manejo de las enfermedades hereditarias de la retina y la coroides. 2a ed. Madrid: MacLine S.L, 2013.
21. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006; 1: 40.
22. Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol 2012; 25: 42–49.
23. Renner AB, Kellner U, Cropp E, et al. Choroideremia: variability of clinical and electrophysiological characteristics and first report of a negative electroretinogram. Ophthalmology 2006; 113: 2066.e1–10.
24. Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis 2007; 2: 7.
26. Fazzi E, Signorini SG, Scelsa B, et al. Leber's congenital amaurosis: An update. Eur J Paediatr Neurol 2003; 7: 13–22.
28. Rubin M. Choroideremia: study of a family and literature review. Arch Ophtalmol 1966; 76: 563–574.
29. Mura M, Sereda C, Jablonski M, et al. Clinical and functional finding in choroideremia due to complete deletion of the CHM gene. Arch Ophtalmol 2007; 125: 1109–1113.
30. Lyon M. Gene action in X chromosome of mouse. Nature 1996; 190: 372–373.
31. Cheung M, Nune G, Wang M, et al. Detection of Localized Retinal Dysfunction in a Choroideremia Carrier. Am J Ophthalmol 2004; 415: 189–191.
32. Furgoch M, Mewes-Arès J, Radziwon A, et al. Molecular genetic diagnostic techniques in choroideremia. Mol Vis 2014; 20: 535–544.
33. Wilson D, Weleber R, Green W. Ocular clinicopathologic study of gyrate atrophy. Am J Ophthalmol 1991; 111: 24–33.
34. Hayasaka S, Shoji K, Kanno C. Differential diagnosis of diffuse choroidal atrophies, diffuse choriocapillaris atrophy, choroideremia, and gyrate atrophy of the choroid and retina. Retina 1985; 5: 30–37.
35. Kurstjens J. Choroideremia and Gyrate atrophy of the choroid and retina. Doc Ophthalmol 1965; 19: 1.
36. Takki K. Gyrate atrophy of the choroid and retina with hyperornithinemia. Br J Ophthalmol 1974; 58: 53.
38. Carr R. Congenital stationary nightblindness. Trans Am Ophthalmol Soc 1974; LXXII: 448–487.
39. Watanabe I, Taniguchi Y, Morioka K, et al. Congenital stationary night blindness with myopia: a clinico-pathologic study. Doc Ophthalmol 1986; 63: 55–62.
40. Strom TM, Nyakatura G, Apfelstedt-sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet 1998; 19: 260–263.
41. Rao V, Cohen G, Oprian D. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature 1994; 367: 639–642.
42. Ripps H. Night blindness revisited: from man to molecules. Invest Ophthalmol Vis Sci 1982; 23: 588–609.
43. Dryja T, Hahn L, Reboul T, et al. Missense mutation in the gene encoding the a subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet 1996; 13: 358–360.
44. Miyake Y, Yagasaki K, Horiguchi M, et al. Congenital Stationary Night Blindness With Negative Electroretinogram. Arch Ophtalmol 1996; 104: 1013–1020.
45. Simunovic M. The cone dystrophies. Eye 1998; 12: 553–565.
46. Thiadens A a HJ, Phan TML, Zekveld-Vroon RC, et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012; 119: 819– 826.
56. Gamundi MJ, Hernan I, Muntanyola M, et al. High prevalence of mutations in peripherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Mol Vis 2007; 13: 1031–1037.
60. Boon CJF, Van Den Born LI, Visser L, et al. Autosomal recessive bestrophinopathy: Differential diagnosis and treatment options. Ophthalmology 2013; 120: 809–820.
66. Kim DY, Mukai S. X-linked juvenile retinoschisis (XLRS): a review of genotypephenotype relationships. Semin Ophthalmol 2013; 28: 392–396.
70. Liutkeviciene R, Lesauskaite V, Ašmoniene V, et al. Inherited macular dystrophies and differential diagnostics. Medicina (B Aires) 2012; 48: 485–495.
71. Bravo-Gil N, Méndez-Vidal C, Romero-Pérez L, et al. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep 2016; 6: 23910.
72. Molday RS, Kellner U, Weber BHF. X-l inked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2013; 31: 195–212.
73. Kellner U. Imaging and the perspective of clinical electrophysiology. Doc Ophthalmol 2008; 116: 75–77.
74. Kiser AK, Mladenovich D, Eshraghi F, et al. Reliability and consistency of visual acuity and contrast sensitivity measures in advanced eye disease. Optom Vis Sci 2005; 82: 946– 954.
75. Gerth C, Wright T, Heón E, et al. Assessment of Central Retinal Function in Patients with Advanced Retinitis Pigmentosa. Investig Opthalmology Vis Sci 2007; 48: 1312– 1318.
76. Moloney TP, O'Hagan S, Lee L. Ultrawide-field fundus photography of the first reported case of gyrate atrophy from Australia. Clin Ophthalmol 2014; 8: 1561–3.
77. Furino C, Boscia F, Cardascia N, et al. Fundus autofluorescence, optical coherence tomography and visual acuity in adult-onset foveomacular dystrophy. Ophthalmologica 2008; 222: 240–244.
78. Sunness JS, Steiner JN. Retinal function and loss of autofluorescence in stargardt disease. Retina 2008; 28: 794–800.
79. Oishi A, Ogino K, Makiyama Y, et al. Wide-Field Fundus Autofluorescence Imaging of Retinitis Pigmentosa. Ophthalmology 2013; 120: 1827–1834.
80. Ogura S, Yasukawa T, Kato A, et al. Wide-field fundus autofluorescence imaging to evaluate retinal function in patients with retinitis pigmentosa. Am J Ophthalmol 2014; 158: 1093–8.
81. Mitamura Y, Mitamura-Aizawa S, Nagasawa T, et al. Diagnostic imaging in patients with retinitis pigmentosa. J Med Investig 2012; 59: 1–11.
82. Wabbels B, Demmler A, Paunescu K, et al. Fundus autofluorescence in children and teenagers with hereditary retinal diseases. Graefe's Arch Clin Exp Ophthalmol 2006; 244: 36–45.
83. Hajali M, Fishman G a, Anderson RJ. The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. Br J Ophthalmol 2008; 92: 1065–1068.
84. Oh KT, Weleber RG, Stone EM, et al. Electroretinographic findings in patients with Stargardt disease and fundus flavimaculatus. Retina 2004; 24: 920–928.
85. Iijima H, Yamaguchi S, Kogure S, et al. Electroretinogram in cone dystrophy. Jpn J Ophthalmol 1991; 35: 453–466.
86. Kelly JP, Crognale M a., Weiss AH. ERGs, cone-isolating VEPs and analytical techniques in children with cone dysfunction syndromes. Doc Ophthalmol 2003; 106: 289–304.
87. Makiyama Y, Ooto S, Hangai M, et al. Macular Cone Abnormalities in Retinitis Pigmentosa with Preserved Central Vision Using Adaptive Optics Scanning Laser Ophthalmoscopy. PLoS One 2013; 8: e79447.
88. Tojo N, Nakamura T, Fuchizawa C, et al. Adaptive optics fundus images of cone photoreceptors in the macula of patients with retinitis pigmentosa. Clin Ophthalmol 2013; 7: 203–10.
89. González-del Pozo M, Borrego S, Barragán I, et al. Mutation screening of multiple genes in Spanish patients with autosomal recessive retinitis pigmentosa by targeted resequencing. PLoS One 2011; 6: e27894.
90. Akinci A, Oner O, Aktas Z, et al. Refractive errors and strabismus in children with laurence-moon-biedl syndrome. J Pediatr Ophthalmol Strabismus 2009; 47: 26–28.
91. Iannaccone A, Kritchevsky SB, Ciccarelli ML, et al. Kinetics of visual field loss in Usher syndrome Type II. Invest Ophthalmol Vis Sci 2004; 45: 784–792.
92. Young NM, Mets MB, Hain TC. Early diagnosis of Usher syndrome in infants and children. American Journal of Otology 1996; 17: 30–34.
93. McCulloch DL, Marmor MF, Brigell M, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015; 130: 1–12.
94. Odom JV, Bach M, Brigell M, et al. ISCEV standard for clinical visual evoked potentials (2009 update). Doc Ophthalmol 2010; 120: 111–119.
95. Hood DC, Bach M, Brigell M, et al. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc Ophthalmol 2012; 124: 1–13.
96. Marmor MF, Brigell MG, McCulloch DL, et al. ISCEV standard for clinical electrooculography (2010 update). Doc Ophthalmol 2011; 122: 1–7.
97. Bach M, Brigell MG, Hawlina M, et al. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc Ophthalmol 2013; 126: 1–7.
98. Català J, Castany M. Pruebas electrofisiológicas: ¿qué, cuándo, cómo y por qué? (2a parte). Ann d'Oftalmologia 2005; 13: 76–90.
99. Catala J, Castany M. Pruebas electrofisiológicas: ¿qué, cuándo, cómo y por qué? Ann d'Oftalmologia 2005; 13: 8–29.
100. Brecelj J. Visual electrophysiology in the clinical evaluation of optic neuritis, chiasmal tumours, achiasmia, and ocular albinism: an overview. Doc Ophthalmol 2014; 129: 71– 84.
101. Fishman GA, Birch DG, Holder GE, et al. Electrophysiologic testing in disorders of the retina, optic nerve, and visual pathway. 2 edition. San Francisco: Oxford University Press, 2001.
102. Espinos C, Pérez-Garrigues H, Beneyto M, et al. Sordera hereditaria sindrómica: Síndrome de Usher. An Otorrinolaringol Ibero Am 1999; XXVI: 83–95.
103. Calzada D, Vilela C, Vallet M, et al. Protocolo para la exploración neurofisiológica de los pacientes de retinosis pigmentaria incluidos en los estudios de genética molecular. Rev Neurofisiología Clínica 1993; 6: 157–60.
104. Kriss A, Jeffrey B, Taylor D. The electroretinogram in infants and young children. J Clin Neurophysiol 1992; 9: 373–93.
105. Alonso L, Grimaldos P, Boix J, et al. Study of families with retinitis pigmentosa in the geographic area of valencia (Spain). In: 6th World RP Congress-IRPA-CRC. Press London, 1991, pp. 1–8.
106. Millán J, Martínez F, Vilela C, et al. An autosomal dominant retinitis pigmentosa family with close linkage to D7S480 on 7q. Hum Genet 1995; 96: 216–8.
107. Nájera C, Millán JM, Vilela C, et al. Classification of retinitis pigmentosa in a spanish population on the basis of clinical features and mode of inheritance. Genetic.
108. Burguera J, Vilela C, Vallet M, et al. ERG y PEV en pacientes con la enfermedad de parkinson. Arch Neurobiol 1990; 53: 1–7.
109. Burguera J, Vilela C. The electroretinogram and visual evoked potential in parkinson's disease. Park Dig 1991; 4: 3–4.
110. Heckenlively JR, G.B.Arden, Nusinowitz S, et al. Principles Practice Clinical Electrophysiology Vision. Masachusetts, London: The Mit Press Cambridge, 2006.
111. Belda J, Romá J, Vilela C, et al. Serum vitamin e levels negatively correlate with severity of age-related macular degeneration. Med Aging Dev.
112. Hawlina M, Konec B. New noncorneal HK-loop electrode for clinical electroretinography. Doc Ophthalmol 1992; 81: 253–9.
113. Marmor MF, Kellner U, Lai TYY, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology 2011; 118: 415–422.
114. Bergholz R, Rüther K, Schroeter J, et al. Influence of chloroquine intake on the multifocal electroretinogram in patients with and without maculopathy. Doc Ophthalmol 2015; 130: 211–219.
115. Arden BYGB, Kelsey a DJH. Changes Produced By Light in the Standing. J Physiol 1962; 161: 189–204.
116. Arden GB, Barrada. A. An analysis of the electroculograms of a series of normal subjects. Br J Ophthalmol 1962; 46: 468–82.
117. Arden G, Barrada a, Kelsey J. New clinical test of retinal function based upon the standing potential of the eye. Br J Ophthalmol 1962; 46: 449–467.
118. Koenekoop RK, Lopez I, Den Hollander AI, et al. Genetic testing for retinal dystrophies and dysfunctions: Benefits, dilemmas and solutions. Clin Exp Ophthalmol 2007; 35: 473–485.
119. Lambertus S, Lindner M, Bax NM, et al. Progression of Late-Onset Stargardt Disease. Investig Opthalmology Vis Sci 2016; 57: 5186.
120. Walia S, Fishman G a. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet 2009; 30: 63–68.
121. Fishman G. Fundus flavimaculatus. A clinical classification. Arch Ophthalmol 1976; 94: 2061–67.
122. Van Huet R a C, Bax NM, Westeneng-Van Haaften SC, et al. Foveal sparing in Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 7467–7478.
123. Budiene B, Liutkeviciene R, Zaliuniene D. Best vitelliform macular dystrophy: literature review. Open Med 2014; 784–795.
124. North V, Gelman R, Tsang SH. Juvenile-onset macular degeneration and allied disorders. Dev Ophthalmol 2014; 53: 44–52.
125. Querques G, Zerbib J, Georges A, et al. Multimodal analysis of the progression of Best vitelliform macular dystrophy. Mol Vis 2014; 20: 575–592.
126. Ander G, Gorin MB, Polkinghorne PJ, et al. Detection of the Carrier State of X-Linked Retinoschisis. Am J Ophthalmol 1988; 105: 590–595.
127. Bowles K, Cukras C, Turriff A, et al. X-linked retinoschisis: RS1 mutation severity and age affect the ERG phenotype in a cohort of 68 affected male subjects. Invest Ophthalmol Vis Sci 2011; 52: 9250–9256.
128. Macdonald I, Smaoui N, Seabra M. Choroideremia. In: Pagon R, Adam M, Ardinger H (eds) GeneReviews. Seatle (WA): University of Washington, 2014, pp. 1–12.
129. De Laey J. Leber's congenital amaurosis. Bull Soc Belge Ophtalmol 1991; 241: 41–50.
130. Seabra M, Brown M, Goldstein J. Retinal degeneration in choroideremia : Deficiency of Rab ge. Science (80- ) 1993; 259: 377–381.
131. Iannaccone A, Vingolo EM, Rispoli E, et al. Electroretinographic alterations in the Laurence-Moon-Bardet-Biedl phenotype. Acta Ophthalmol Scand 1996; 74: 8–13.
132. Smith RJ, Berlin CI, Hejtmancik JF, et al. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet 1994; 50: 32–38.
133. Pakarinen L, Tuppurainen K, Laippala P, et al. The ophthalmological course of Usher syndrome type III. Int Ophthalmol 1996; 19: 307–311.
134. Mets MB, Young NM, Pass a, et al. Early diagnosis of Usher syndrome in children. Trans Am Ophthalmol Soc 2000; 98: 237–242; discussion 243–245.
135. Tamayo ML, Maldonado C, Plaza SL, et al. Neuroradiology and clinical aspects of Usher syndrome. Clin Genet 1996; 50: 126–132.
136. Abraham FA, Cohen D, Sohmer H. Usher's syndrome: electrophysiological tests of the visual and auditory systems. Doc Ophthalmol 1977; 44: 435–44.
137. Fleischhauer J, Njoh WA, Niemeyer G. Syndromic retinitis pigmentosa: ERG and phenotypic changes. Klin Monbl Augenheilkd 2005; 222: 186–190.
138. Beales P, Elcioglu N, Woolf A, et al. New criteria for improved diagnosis of Bardet- Biedl syndrome: results of a population survey. 1999; 437–446.
139. Saleh SI, Mady S a., Al-Ghanem MM, et al. Bardet-Biedl syndrome: Clinical, endocrinological and imaging evaluation of three unrelated Bedouin children. Med Princ Pract 1998; 7: 230–236.
141. Dammeyer J. Development and characteristics of children with Usher syndrome and CHARGE syndrome. Int J Pediatr Otorhinolaryngol 2012; 76: 1292–1296.
142. Bande M, Pose S, Treus A, et al. Síndrome de Kearns-Sayre : manifestaciones oftalmológicas. Ann Pediatr 2015; 82: e151–3.
143. Finsterer J, Frank M. Diabetes in Kearns-Sayre Syndrome: More Common than Anticipated. Can J Diabetes 2015; 39: 253.
144. Esteves F, Dolz-Marco R, Hernández-Martínez P, et al. Pattern dystrophy of the macula in a case of steinert disease. Case Rep Ophthalmol 2013; 4: 129–33.
145. Kimizuka Y, Kiyosawa M, Tamai M, et al. Retinal changes in myotonic dystrophy. Clinical and follow-up evaluation. Retina 1993; 13: 1129–35.
146. Sigesmund D, Weleber R, Pillers D, et al. Characterization of the ocular phenotype of Duchenne and Becker muscular dystrophy. Ophthalmology 1994; 101: 856–65.
147. Crane D. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem Int 2014; 69: 1–8.
148. Folz S, Trobe J. The peroxisome and the eye. Surv Ophthalmol 1991; 35: 353–68.
149. Steinberg S, Raymond G, Braverman N, et al. Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum. In: Pagon R, Adam M, Ardinger H (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington http://www.ncbi.nlm.nih.gov/books/NBK1448/.
150. Rinaldi E, Cotticelli L, Di Meo A, et al. Ocular findings in Refsum's disease. Metab Pediatr Ophthalmol 1981; 5: 149–54.
151. Grant C a, Berson EL. Treatable forms of retinitis pigmentosa associated with systemic neurological disorders. Int Ophthalmol Clin 2001; 41: 103–10.
152. Hooper A, Burnett J. Update on primary hypobetalipoproteinemia. Curr Atheroscler Rep 2014; 16: 423.
153. Lee J, Hegele R a. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: A framework for diagnosis and management. J Inherit Metab Dis 2014; 37: 333–339.
154. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014; 25: 161–8.
155. Alshareef R a., Bansal AS, Chiang A, et al. Macular atrophy in a case of abetalipoproteinemia as only ocular clinical feature. Can J Ophthalmol / J Can d'Ophtalmologie 2015; 50: e43–e46.
156. Wong A, Héon E. Helicoid peripapillary chorioretinal degeneration in abetalipoproteinemia. Arch Ophthalmol 1998; 116: 250–1.
157. Duker J, Belmont J, Bosley T. Angioid streaks associated with abetalipoproteinemia. Case report. Arch Ophthalmol 1987; 105: 1173–4.
158. Sperling MA, Hiles DA, Kennerdell JS. Electroretinographic responses following vitamin A therapy in A-beta-lipoproteinemia. Am J Ophthalmol 1972; 73: 342–51.
159. Sirisena ND, Thalgahagoda S, Abeyagunawardena A, et al. Novel COL4A3 gene mutations in a consanguineous family with autosomal recessive Alport syndrome. Nephrology 2015; 20: 580–580.
160. Savige J, Sheth S, Leys A, et al. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol 2015; 10: 703–709.
161. Zylbermann R, Silverstone B, Brandes E, et al. Retinal lesions in Alport's syndrome. J Pediatr Ophthalmol Strabismus 1980; 17: 255–60.
162. Sohar E. A heredo-familial syndrome characterized by renal disease, inner ear deafness, and ocular changes. Harefuah 1954; 47: 161–2.
163. Sjögren T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorders; a clinical and genetic study. Acta Psychiatr Neurol Scand 1957; 113: 1–112.
164. Sanabria MR, Coco RM. Sjögren-Larsson Syndrome. Ophthalmology 2011; 118: 2101– 2102.
165. Lund C, Bjørnvold M, Tuft M, et al. Aicardi syndrome: an epidemiologic and clinical study in Norway. Pediatr Neurol 2015; 52: 182–6.e3.
166. Martel JN, Rutar T, Lujan BJ, et al. Chorioretinal architecture in Aicardi syndrome: an optical coherence tomography and fluorescein angiography study. J AAPOS 2011; 15: 308–10.
167. Guerriero S, Sciruicchio V, De Blasi R, et al. Chorioretinal lacunae: pathognomonic findings for Aicardi syndrome. J Pediatr Ophthalmol Strabismus 2010; 21: e1–3.
168. Nowak V, Bremner F, Massey L, et al. Kjellin syndrome: hereditary spastic paraplegia with pathognomonic macular appearance. Pr Neurol 2014; 14: 278–9.
169. Farmer S, Longstreth WJ, Kalina R, et al. Fleck retina in Kjellin's syndrome. Am J Ophthalmol 1985; 99: 45–50.
170. Tarantola RM, Nguyen V, Agarwal A. Characterization of Kjellin Syndrome using Spectral-Domain Optical Coherence Tomography and Fundus Autofluorescence. Retin Cases Br Reports 2011; 5: 49–55.
171. Puech B, Lacour A, Stevanin G, et al. Kjellin syndrome: long-term neuroophthalmologic follow-up and novel mutations in the SPG11 gene. Ophthalmology 2011; 118: 564–73.
172. Suzuki T, Miyake N, Tsurusaki Y, Okamoto N, Alkindy A, Inaba A, Sato M, Ito S, Muramatsu K, Kimura S, Ieda D, Saitoh S, Hiyane M, Suzumura H, Yagyu K, Shiraishi H, Nakajima M, Fueki N, Habata Y, Ueda Y, Komatsu Y, Yan K, Shimoda K, Shitara Y, Mizuno S, Ic MN. Molecular genetic analysis of 30 families with Joubert syndrome. Clin Genet 2016; 1–11.
173. Adams N a, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet 2007; 28: 113–25.
174. Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010; 5: 20.
175. Valente E, Dallapiccola B, Bertini E. Joubert syndrome and related disorders. Handb Clin Neurol 2013; 113: 1879–88.
176. Knopp C, Rudnik-Schöneborn S, Eggermann T, et al. Syndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing. Mol Cell Probes 2015; 29: 299–307.
177. Álvarez-Satta M, Castro-sánchez S, Valverde D. Alström syndrome : current perspectives. Appl Clin Genet 2015; 8: 171–179.
178. Stone E, Ho CY. Genetic testing for inherited heart disease. Arch Ophtalmol 2007; 125: 205–212.
179. O'Sullivan J, Mullaney BG, Bhaskar SS, et al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet 2012; 49: 322–326.
180. Heon E. Predictive DNA testing in ophthalmology: View 1. Br J Ophthalmol 2003; 87: 633–635.
181. MacDonald IM, Tran M, Musarella MA. Ocular genetics: current understanding. Surv Ophthalmol 2004; 49: 159–196.
182. Ministerios de Sanidad SS e I. Orden SSI/2065/2014, de 31 de octubre, por la que se modifican los anexos I, II y III del Real Decreto 1030/2006, de 15 de septiembre, por el que se establece la cartera de servicios comunes del Sistema Nacional de Salud y el procedimiento para su actuali. 2014.
183. Alapati A, Goetz K, Suk J, et al. Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Invest Ophthalmol Vis Sci 2004; 31: 15510–5521.
184. Combs R, McAllister M, Payne K, et al. Understanding the impact of genetic testing for inherited retinal dystrophy. Eur J Hum Genet 2013; 21: 1–5.
185. Eden M, Payne K, Combs RM, et al. Valuing the benefits of genetic testing for retinitis pigmentosa: a pilot application of the contingent valuation method. Br J Ophthalmol 2013; 97: 1051–1056.
186. Mackey D, Heon E, Webster A. Predictive DNA testing in ophthalmology: View 2. Br J Ophthalmol 2003; 87: 633.
187. Wiggs J, Pierce E. Genetic testing for inherited eye disease: who benefits? JAMA Ophthalmol 2013; 131: 9–10.
188. Sutherland JE, Day M a. Advantages and disadvantages of molecular testing in ophthalmology. Expert Rev Ophthalmol 2011; 6: 221–245.
189. Zanolli MT, Khetan V, Dotan G, et al. Should patients with ocular genetic disorders have genetic testing? Curr Opin Ophthalmol 2014; 25: 359–365.
190. Stroh E. Taking the family history in genetic disease. Curr Opin Ophthalmol 2011; 22: 340–346.
191. Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res 2010; 29: 335–375.
192. Downs K, Zacks DN, Caruso R, et al. Molecular testing for hereditary retinal disease as part of clinical care. Arch Ophthalmol 2007; 125: 252–258.
193. Wang DY, Chan WM, Tam POS, et al. Gene mutations in retinitis pigmentosa and their clinical implications. Clin Chim Acta 2005; 351: 5–16.
194. Fan BJ, Tam POS, Choy KW, et al. Molecular diagnostics of genetic eye diseases. Clin Biochem 2006; 39: 231–239.
195. Evans K, Gregory CY, Er AFRY, et al. The role of molecular genetics in the prenatal diagnosis of retina l dystrophies. Eye 1995; 9: 24–28.
196. Wang F, Wang H, Tuan H-F, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet 2014; 133: 331–345.
197. Corton M, Nishiguchi KM, Avila-Fernández A, et al. Exome Sequencing of Index Patients with Retinal Dystrophies as a Tool for Molecular Diagnosis. PLoS One 2013; 8: e65574.
198. Fu Q, Wang F, Wang H, et al. Next-Generation Sequencing–Based Molecular Diagnosis of a Chinese Patient Cohort With Autosomal Recessive Retinitis Pigmentosa. Investig Opthalmology Vis Sci 2013; 54: 4158.
199. Golovleva I, Köhn L, Burstedt M, et al. Retinal Degenerative Diseases. 2010; 664: 1–6.
200. Li S, Shen T, Xiao X, et al. Detection of CRB1 mutations in families with retinal dystrophy through phenotype-oriented mutational screening. Int J Mol Med 2014; 913– 918.
201. Audo I, Bujakowska KM, Leveillard T, et al. Development and application of a nextgeneration- sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis 2012; 7: 8.
202. Thiadens A, Klaver C. Genetic Testing and Clinical Characterization of Patients with Cone-Rod Dystrophy. Invest Ophthalmol Vis Sci 2010; 51: 6904–6905.
203. Fujinami K, Zernant J, Chana RK, et al. ABCA4 Gene Screening by Next-Generation Sequencing in a British Cohort. Investig Opthalmology Vis Sci 2013; 54: 6662.
204. Drenser KA, Dailey W, Vinekar A, et al. Clinical presentation and genetic correlation of patients with mutations affecting the FZD4 gene. Arch Ophthalmol 2009; 127: 1649– 1654.
205. Bhatti MT. Retinitis pigmentosa, pigmentary Retinopathies, and Neurologic Diseases.
206. Jaijo T, Aller E, Beneyto M, et al. Estudio genético molecular del síndrome de Usher en España. 2005; 1: 285–289.
207. McKibbin M, Ahmed M, Allsop MJ, et al. Current understanding of genetics and genetic testing and information needs and preferences of adults with inherited retinal disease. Eur J Hum Genet 2014; 22: 1058–1062.
208. Forsythe E, Beales PL. Bardet–Biedl syndrome. Eur J Hum Genet 2013; 21: 8–13.
209. Scanga HL, Nischal KK. Genetics and Ocular Disorders. Pediatr Clin North Am 2014; 61: 555–565.
210. Wang X, Wang H, Sun V, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet 2013; 50: 674–688.
211. Khan A, Al-Mesfer S, Al-Turkmani S, et al. Genetic analysis of strictly defined Leber congenital amaurosis with (and without) neurodevelopmental delay. Br J Ophthalmol 2014; 98: 1724–1728.
212. Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotypephenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004; 23: 306– 317.
213. Stone E. Leber Congenital Amaurosis–A Model for Efficient Genetic Testing of Heterogeneous Disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol 2007; 144: 791–811.
214. Chan S, Freund PR, MacDonald I. Advances in the genetics of eye diseases. Curr Opin Pulm Med 2013; 25: 645–52.
215. Gillespie RL, Hall G, Black GC. Genetic testing for inherited ocular disease: delivering on the promise at last? Clin Experiment Ophthalmol 2014; 42: 65–77.
216. Mezer E, Sutherland J, Goei SL, et al. Utility of molecular testing for related retinal dystrophies. Can J Ophthalmol / J Can d'Ophtalmologie 2006; 41: 190–196.
217. Pradhan M, Hayes I, Vincent A. An audit of genetic testing in diagnosis of inherited retinal disorders: a prerequisite for gene-specific intervention. Clin Experiment Ophthalmol 2009; 37: 703–711.
218. Goodwin P. Hereditary retinal disease. Curr Opin Ophthalmol 2008; 19: 255–262.
219. Shanks ME, Downes SM, Copley RR, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in earlyonset disease. Eur J Hum Genet 2013; 21: 274–280.
220. Chang S, Vaccarella L, Olatunji S, et al. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genomics 2011; 12: 267–75.
221. Ruddle J. Gene testing for the next generation. Clin Experiment Ophthalmol 2014; 42: 1–1.
222. Sutherland J, Day M. Genetic counseling and genetic testing in ophthalmology. Curr Opin Ophthalmol 2009; 20: 343–350.
223. Ernest PJG, Boon CJF, Klevering BJ, et al. Outcome of ABCA4 microarray screening in routine clinical practice. Mol Vis 2009; 15: 2841–7.
224. Clark GR, Crowe P, Muszynska D, et al. Development of a Diagnostic Genetic Test for Simplex and Autosomal Recessive Retinitis Pigmentosa. Ophthalmology 2010; 117: 2169–2177.e3.
225. Neveling K, Collin RWJ, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012; 33: 963–972.
226. Zaneveld J, Wang F, Wang X, et al. Dawn of ocular gene therapy: Implications for molecular diagnosis in retinal disease. Sci China Life Sci 2013; 56: 125–133.
227. Daiger S, Sullivan L, Bowne S. Genes and mutations causing retinitis pigmentosa. Clin Genet 2013; 84: 1–16.
228. Anasagasti A, Irigoyen C, Barandika O, et al. Current mutation discovery approaches in Retinitis Pigmentosa. Vision Res 2012; 75: 117–129.
229. Willis T, Potrata B, Ahmed M, et al. Understanding of and attitudes to genetic testing for inherited retinal disease: a patient perspective. Br J Ophthalmol 2013; 97: 1148–1154.
230. Bong C, Potrata B, Hewison J, et al. Attitudes of patients and relatives/carers towards genetic testing for inherited retinal disease. Eye (Lond) 2010; 24: 1622–1625.
231. Kimberling WJ, Hildebrand MS, Shearer a E, et al. Frequency of Usher Syndrome in Two Pediatric Populations: Implications for genetic screening of Deaf and Hard of Hearing Children. Genet Med 2011; 12: 512–516.
232. Jefatura del Estado. Ley 14/2007, de 3 de julio, de Investigación biomédica. 2007.
233. Branham K, Yashar BM. Providing comprehensive genetic-based ophthalmic care. Clin Genet 2013; 84: 183–189.
234. Schwartz M, Rosenberg T. Prenatal diagnosis of choroidemia. Acta Ophthalmol Scand 1996; 74: 33–6.
235. Mackey D. -Overview. Predictive DNA testing in ophthalmology. Br J Ophthalmol 2003; 87: 637–8.
236. Potrata B, McKibbin M, Lim JN, et al. 'To perpetuate blindness!': attitudes of UK patients with inherited retinal disease towards genetic testing. J Community Genet 2014; 5: 215–22.
237. Ayuso C, Dal-Ré R. Comunicación de los resultados a los participantes en la investigación genética de las enfermedades raras. In: Ayuso C, Dal-Ré R, Palau F (eds) Ética en la investigación de las enfermedades raras. Madrid: Ergon, 2016, pp. 57–73.


